

Soft protein-protein docking in internal coordinates
- PMID: 11790838
- PMCID: PMC2373434
- DOI: 10.1110/ps.19202
Soft protein-protein docking in internal coordinates
Abstract
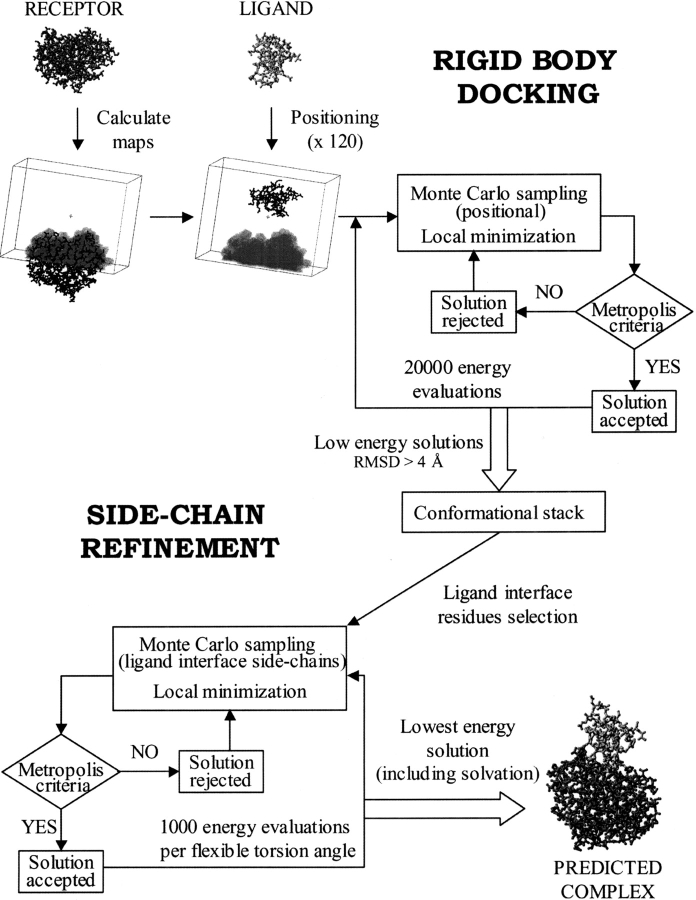
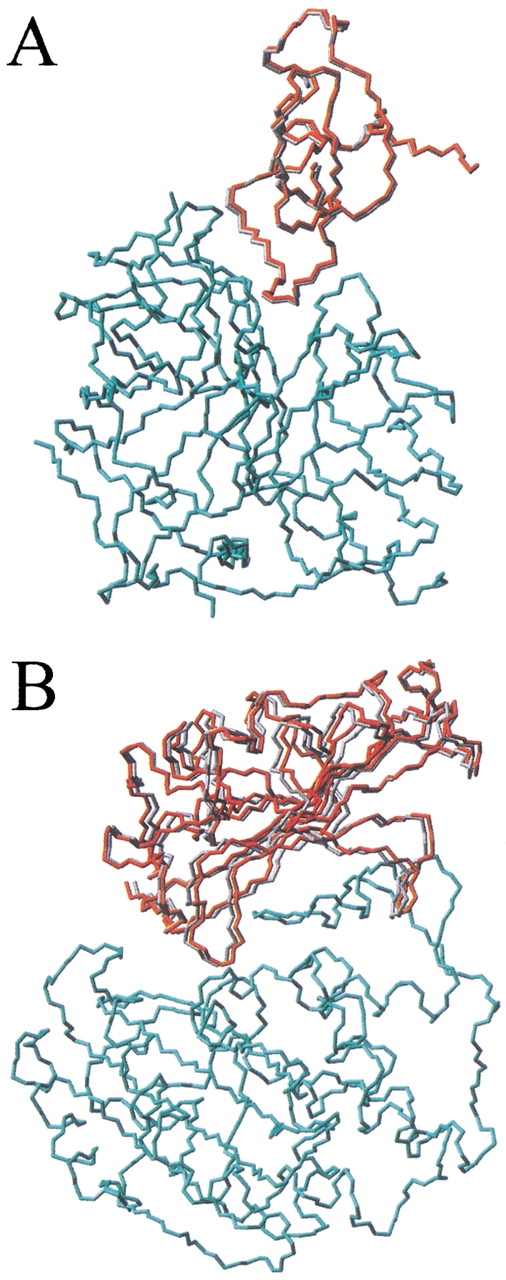
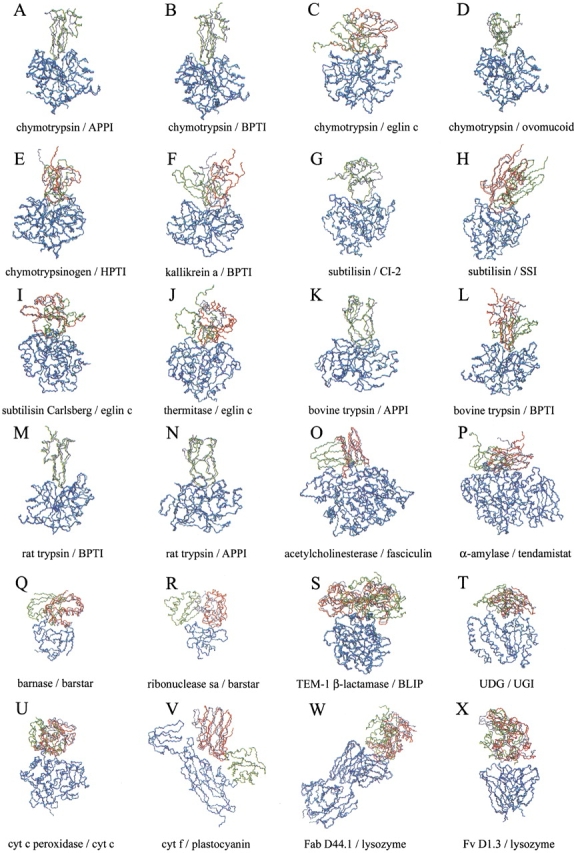
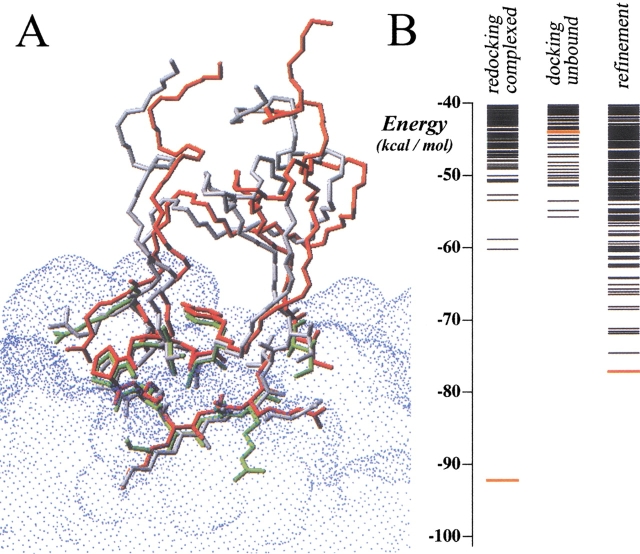
The association of two biological macromolecules is a fundamental biological phenomenon and an unsolved theoretical problem. Docking methods for ab initio prediction of association of two independently determined protein structures usually fail when they are applied to a large set of complexes, mostly because of inaccuracies in the scoring function and/or difficulties on simulating the rearrangement of the interface residues on binding. In this work we present an efficient pseudo-Brownian rigid-body docking procedure followed by Biased Probability Monte Carlo Minimization of the ligand interacting side-chains. The use of a soft interaction energy function precalculated on a grid, instead of the explicit energy, drastically increased the speed of the procedure. The method was tested on a benchmark of 24 protein-protein complexes in which the three-dimensional structures of their subunits (bound and free) were available. The rank of the near-native conformation in a list of candidate docking solutions was <20 in 85% of complexes with no major backbone motion on binding. Among them, as many as 7 out of 11 (64%) protease-inhibitor complexes can be successfully predicted as the highest rank conformations. The presented method can be further refined to include the binding site predictions and applied to the structures generated by the structural proteomics projects. All scripts are available on the Web.
Figures
Similar articles
-
Rapid refinement of protein interfaces incorporating solvation: application to the docking problem.J Mol Biol. 1998 Feb 13;276(1):265-85. doi: 10.1006/jmbi.1997.1519. J Mol Biol. 1998. PMID: 9514726
-
BiGGER: a new (soft) docking algorithm for predicting protein interactions.Proteins. 2000 Jun 1;39(4):372-84. Proteins. 2000. PMID: 10813819
-
RDOCK: refinement of rigid-body protein docking predictions.Proteins. 2003 Nov 15;53(3):693-707. doi: 10.1002/prot.10460. Proteins. 2003. PMID: 14579360
-
Principles of docking: An overview of search algorithms and a guide to scoring functions.Proteins. 2002 Jun 1;47(4):409-43. doi: 10.1002/prot.10115. Proteins. 2002. PMID: 12001221 Review.
-
Recent progress and future directions in protein-protein docking.Curr Protein Pept Sci. 2008 Feb;9(1):1-15. doi: 10.2174/138920308783565741. Curr Protein Pept Sci. 2008. PMID: 18336319 Review.
Cited by
-
Configurational-bias sampling technique for predicting side-chain conformations in proteins.Protein Sci. 2006 Sep;15(9):2029-39. doi: 10.1110/ps.062165906. Protein Sci. 2006. PMID: 16943441 Free PMC article.
-
Computer applications for prediction of protein-protein interactions and rational drug design.Adv Appl Bioinform Chem. 2009;2:101-23. Epub 2009 Nov 10. Adv Appl Bioinform Chem. 2009. PMID: 21918619 Free PMC article.
-
ASPDock: protein-protein docking algorithm using atomic solvation parameters model.BMC Bioinformatics. 2011 Jan 27;12:36. doi: 10.1186/1471-2105-12-36. BMC Bioinformatics. 2011. PMID: 21269517 Free PMC article.
-
Modeling, docking, and fitting of atomic structures to 3D maps from cryo-electron microscopy.Methods Mol Biol. 2013;955:229-41. doi: 10.1007/978-1-62703-176-9_13. Methods Mol Biol. 2013. PMID: 23132064 Free PMC article.
-
Improved side-chain modeling for protein-protein docking.Protein Sci. 2005 May;14(5):1328-39. doi: 10.1110/ps.041222905. Epub 2005 Mar 31. Protein Sci. 2005. PMID: 15802647 Free PMC article.
References
-
- Abagyan, R. and Mazur, A.K. 1989. New methodology for computer-aided modelling of biomolecular structure and dynamics, 2: Local deformations and cycles. J. Biomol. Struct. Dyn. 6 833–845. - PubMed
-
- Abagyan, R. and Argos, P. 1992. Optimal protocol and trajectory visualization for conformational searches of peptides and proteins. J. Mol. Biol. 225 519–532. - PubMed
-
- Abagyan, R. and Totrov, M. 1994. Biased probability Monte Carlo conformational searches and electrostatic calculations for peptides and proteins. J. Mol. Biol. 235 983–1002. - PubMed
-
- Abagyan, R., Totrov, M., and Kuznetsov, D. 1994. ICM: A new method for structure modeling and design: Applications to docking and structure prediction from the distorted native conformation. J. Comp. Chem. 15 488–506.
-
- Ausiello, G., Cesareni, G., and Helmer-Citterich, M. 1997. ESCHER: A new docking procedure applied to the reconstruction of protein tertiary structure. Proteins 28 556–567. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
