

Toward genomic identification of beta-barrel membrane proteins: composition and architecture of known structures
- PMID: 11790840
- PMCID: PMC2373429
- DOI: 10.1110/ps.29402
Toward genomic identification of beta-barrel membrane proteins: composition and architecture of known structures
Abstract
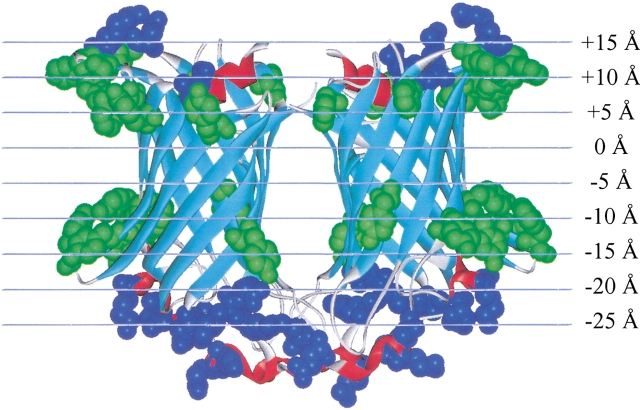
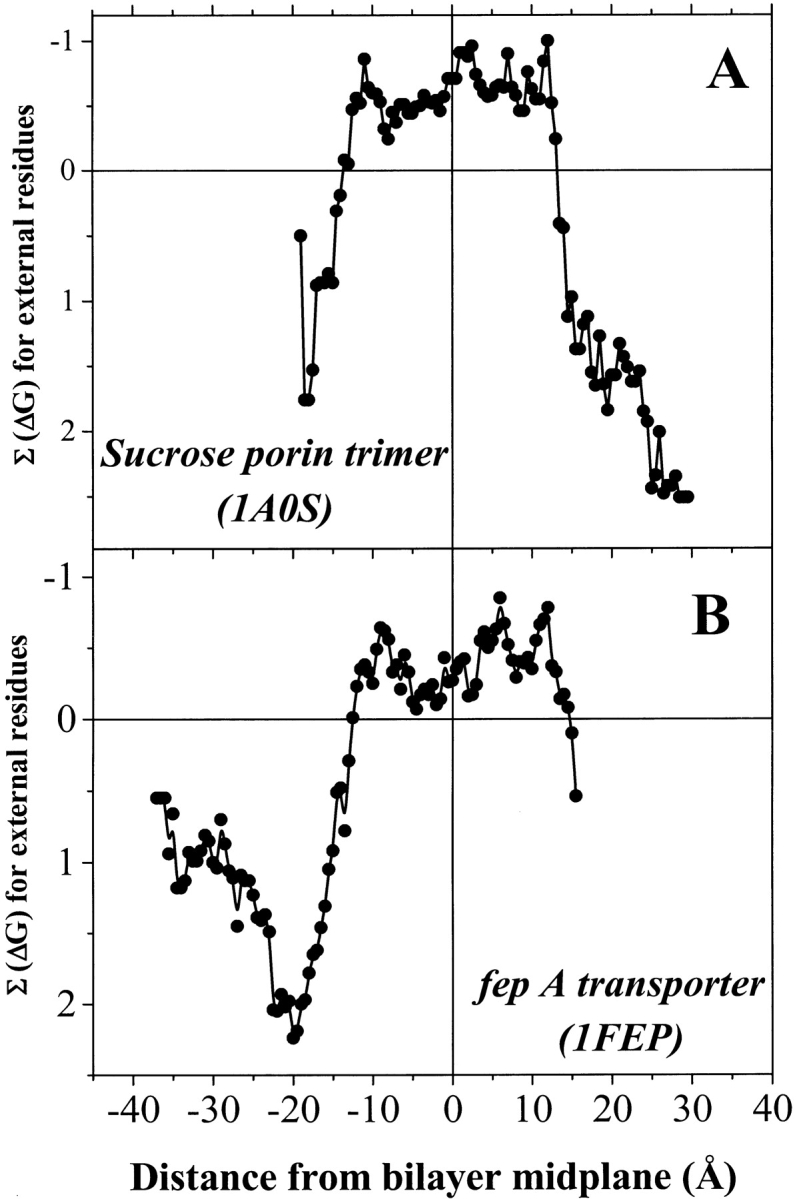
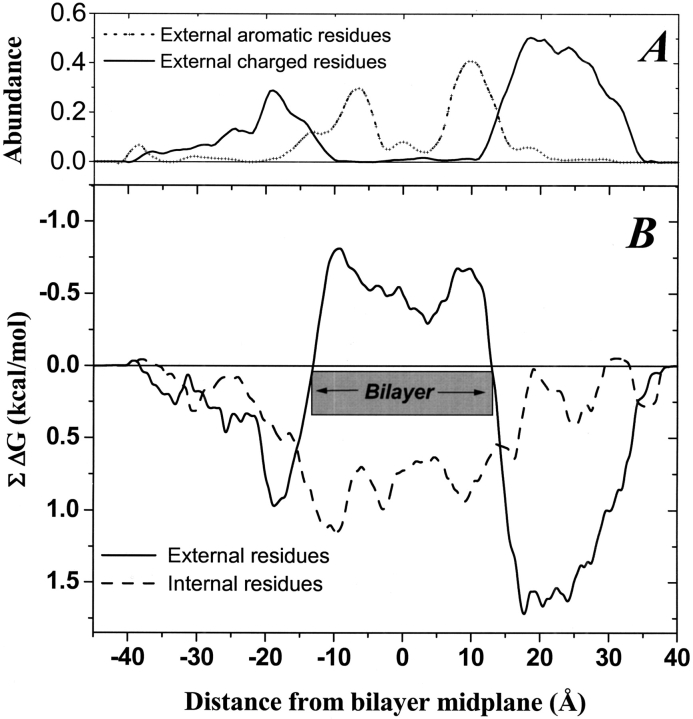
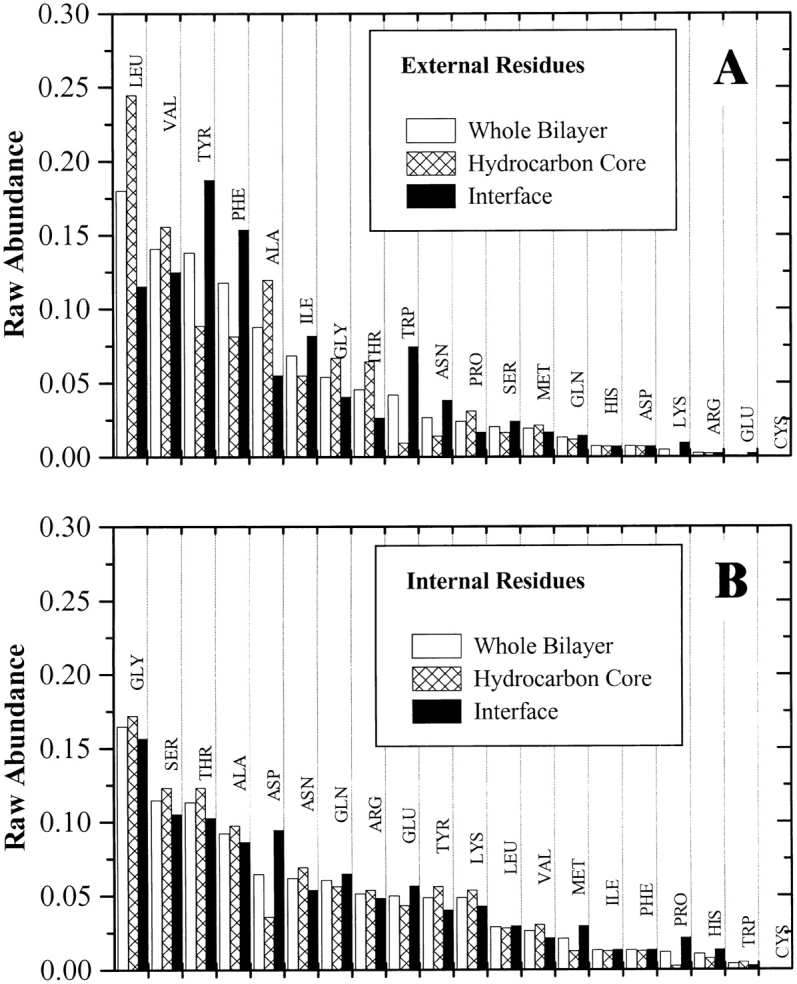
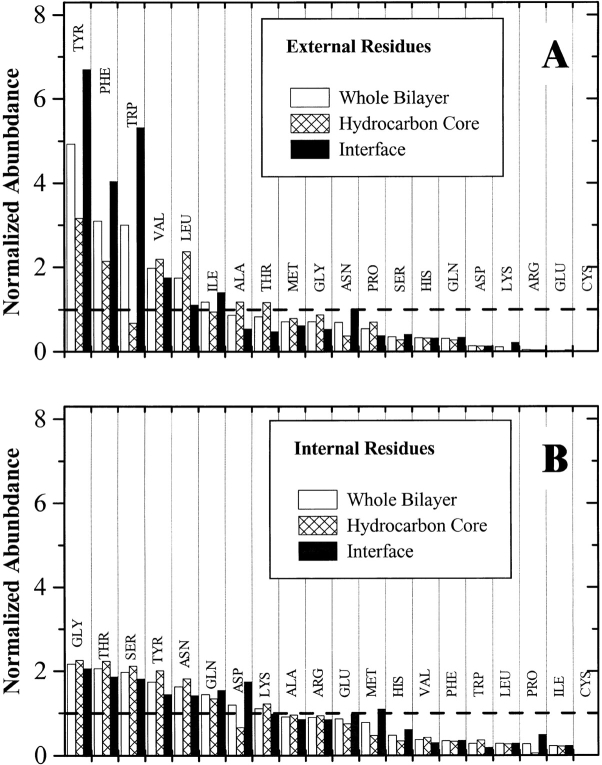
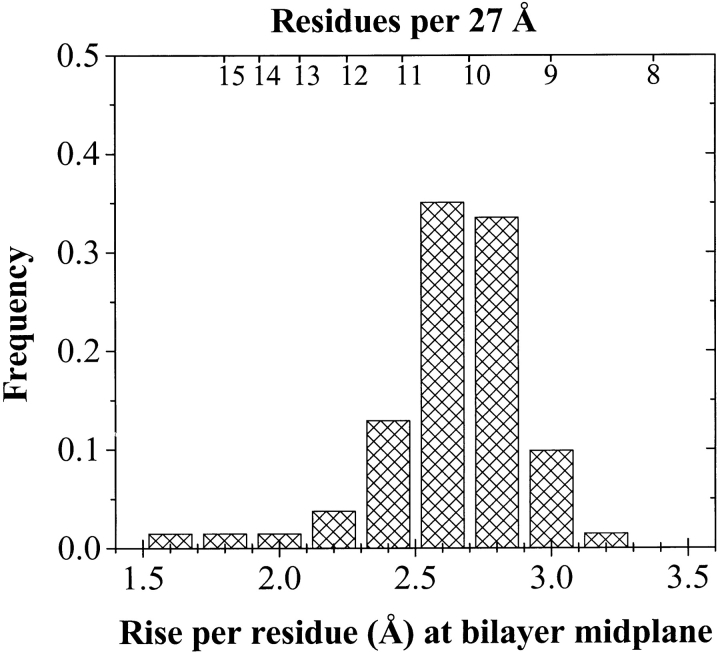
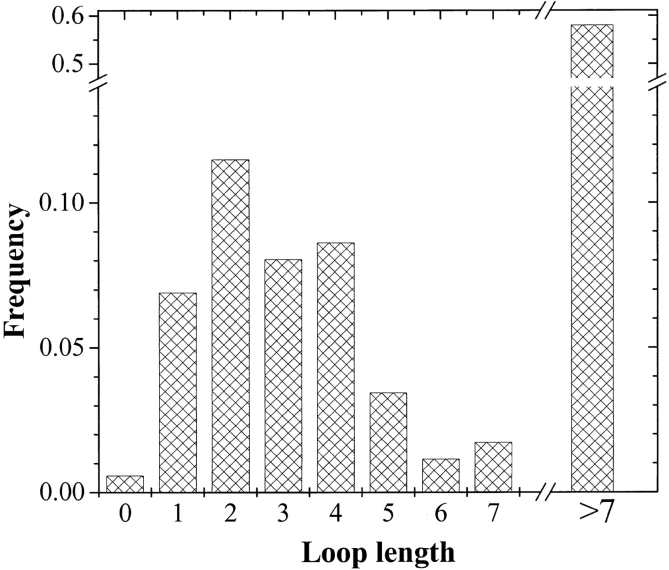
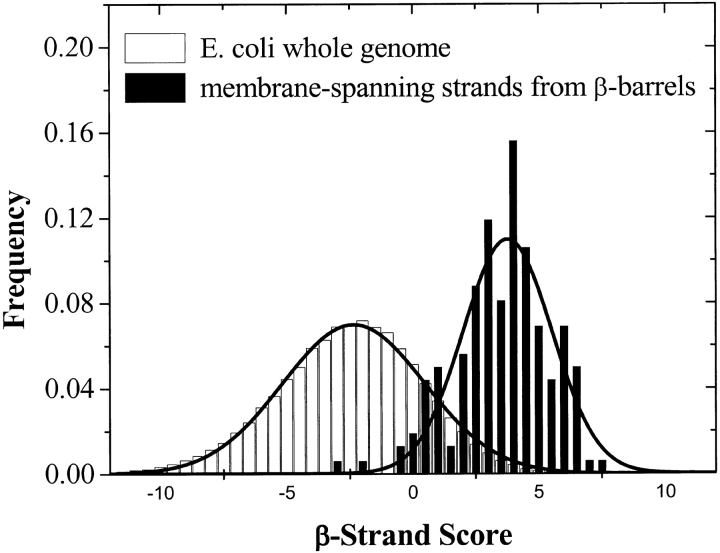
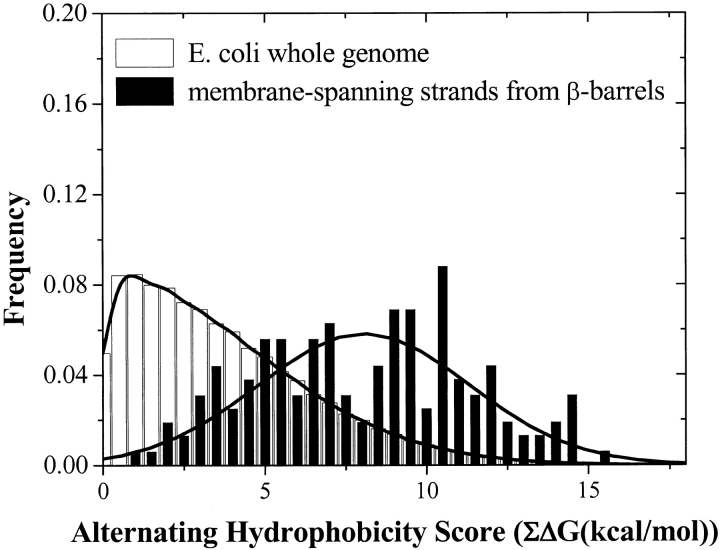
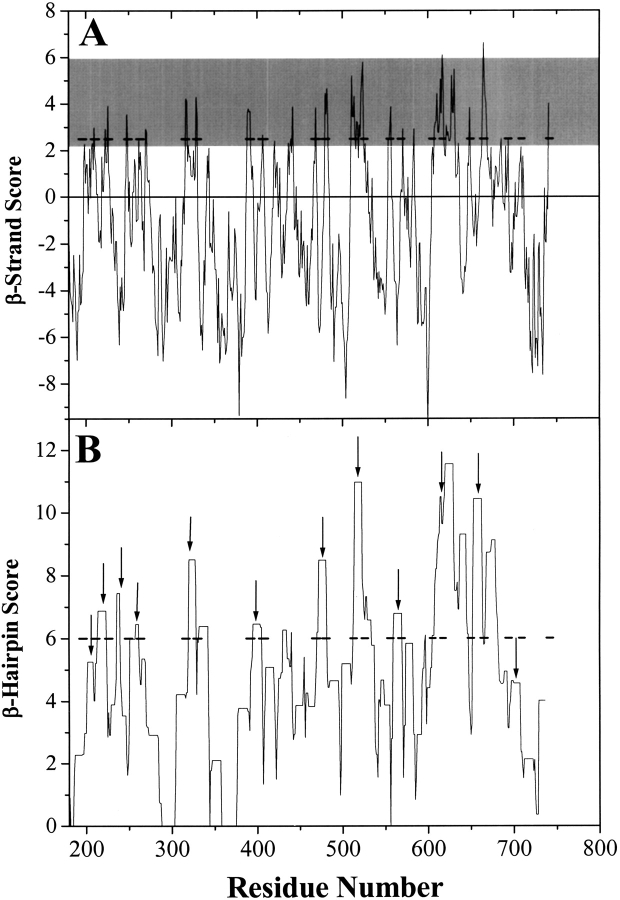
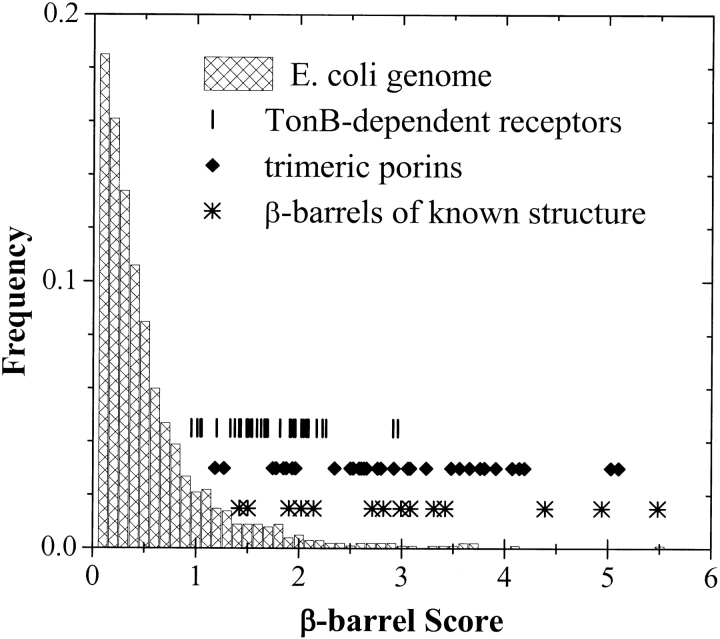
The amino acid composition and architecture of all beta-barrel membrane proteins of known three-dimensional structure have been examined to generate information that will be useful in identifying beta-barrels in genome databases. The database consists of 15 nonredundant structures, including several novel, recent structures. Known structures include monomeric, dimeric, and trimeric beta-barrels with between 8 and 22 membrane-spanning beta-strands each. For this analysis the membrane-interacting surfaces of the beta-barrels were identified with an experimentally derived, whole-residue hydrophobicity scale, and then the barrels were aligned normal to the bilayer and the position of the bilayer midplane was determined for each protein from the hydrophobicity profile. The abundance of each amino acid, relative to the genomic abundance, was calculated for the barrel exterior and interior. The architecture and diversity of known beta-barrels was also examined. For example, the distribution of rise-per-residue values perpendicular to the bilayer plane was found to be 2.7 +/- 0.25 A per residue, or about 10 +/- 1 residues across the membrane. Also, as noted by other authors, nearly every known membrane-spanning beta-barrel strand was found to have a short loop of seven residues or less connecting it to at least one adjacent strand. Using this information we have begun to generate rapid screening algorithms for the identification of beta-barrel membrane proteins in genomic databases. Application of one algorithm to the genomes of Escherichia coli and Pseudomonas aeruginosa confirms its ability to identify beta-barrels, and reveals dozens of unidentified open reading frames that potentially code for beta-barrel outer membrane proteins.
Figures
References
-
- Altschul, S.F., Gish, W., Miller, W., Myers, E.W., and Lipman, D.J. 1990. Basic local alignment search tool. J. Mol. Biol. 215403–410. - PubMed
-
- Benz, R. 1994. Permeation of hydrophilic solutes through mitochondrial outer membranes: Review on mitochondrial porins. Biochim. Biophys. Acta 1197167–196. - PubMed
-
- Bishop, C.M., Walkenhorst, W.F., and Wimley, W.C. 2001. Folding of β-sheet membrane proteins: Specificity and promiscuity in peptide model systems. J. Mol. Biol. 309975–988. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
