

Persistently conserved positions in structurally similar, sequence dissimilar proteins: roles in preserving protein fold and function
- PMID: 11790845
- PMCID: PMC2373454
- DOI: 10.1110/ps.18602
Persistently conserved positions in structurally similar, sequence dissimilar proteins: roles in preserving protein fold and function
Abstract
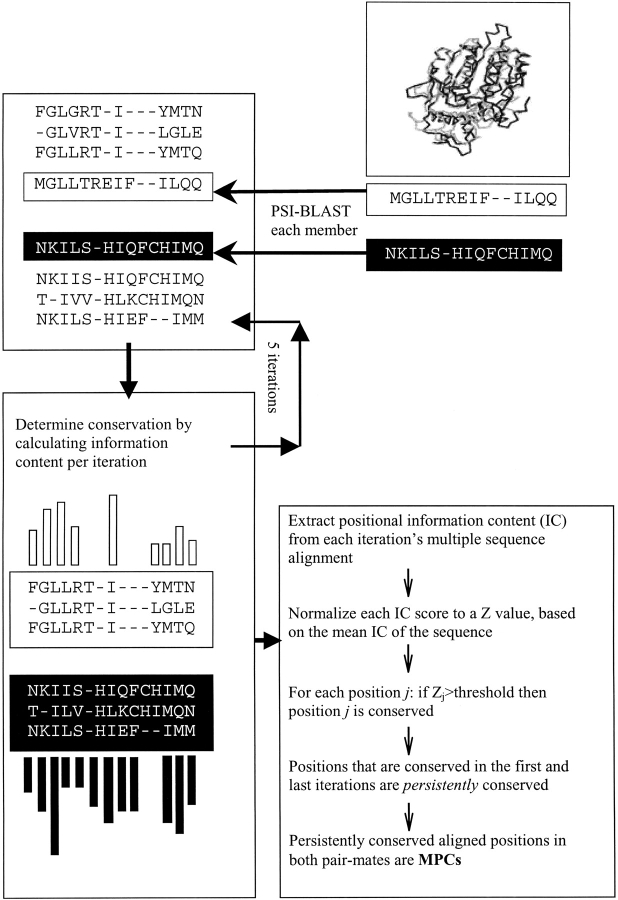
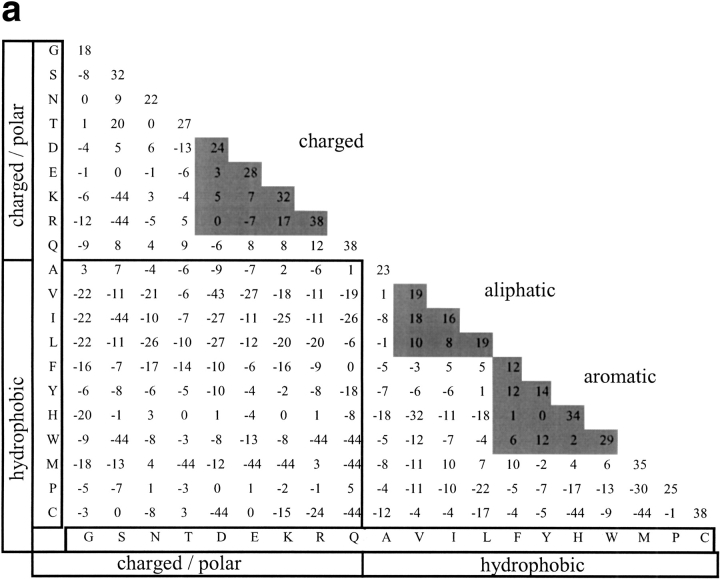
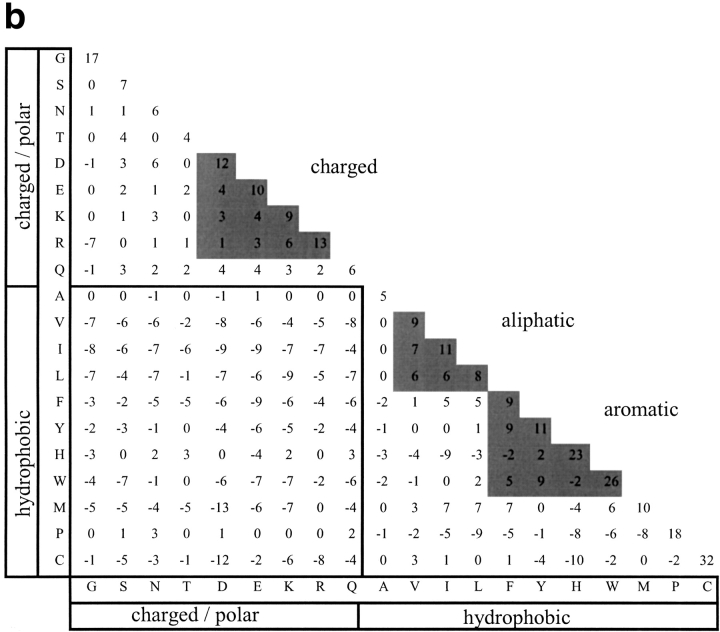
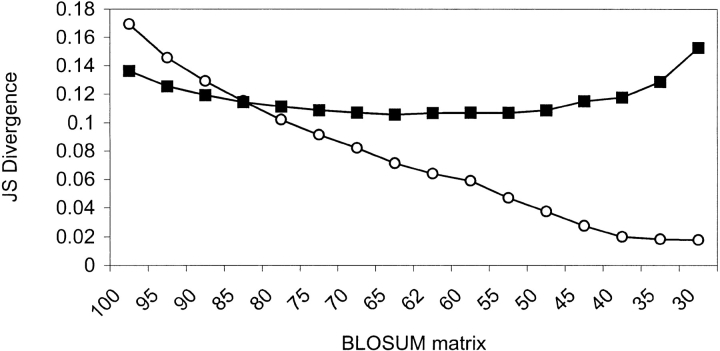
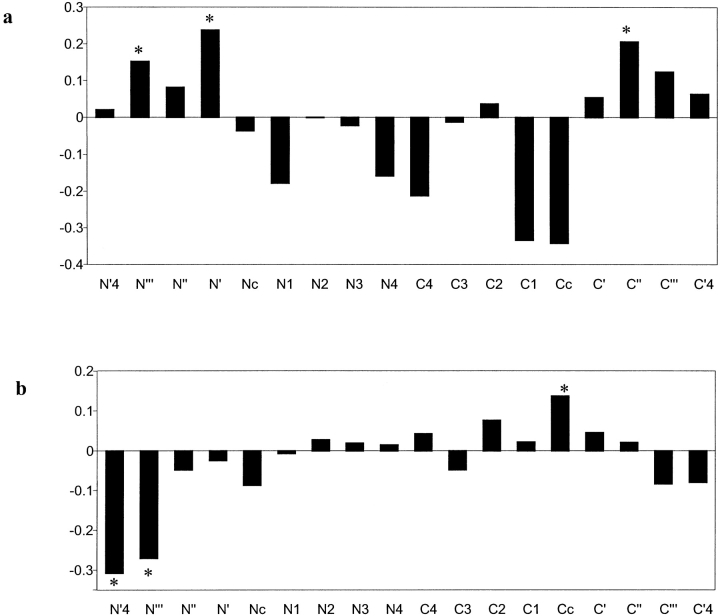
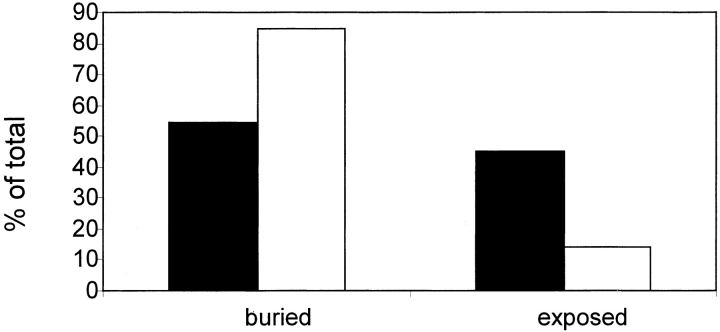
Many protein pairs that share the same fold do not have any detectable sequence similarity, providing a valuable source of information for studying sequence-structure relationship. In this study, we use a stringent data set of structurally similar, sequence-dissimilar protein pairs to characterize residues that may play a role in the determination of protein structure and/or function. For each protein in the database, we identify amino-acid positions that show residue conservation within both close and distant family members. These positions are termed "persistently conserved". We then proceed to determine the "mutually" persistently conserved (MPC) positions: those structurally aligned positions in a protein pair that are persistently conserved in both pair mates. Because of their intra- and interfamily conservation, these positions are good candidates for determining protein fold and function. We find that 45% of the persistently conserved positions are mutually conserved. A significant fraction of them are located in critical positions for secondary structure determination, they are mostly buried, and many of them form spatial clusters within their protein structures. A substitution matrix based on the subset of MPC positions shows two distinct characteristics: (i) it is different from other available matrices, even those that are derived from structural alignments; (ii) its relative entropy is high, emphasizing the special residue restrictions imposed on these positions. Such a substitution matrix should be valuable for protein design experiments.
Figures

References
-
- Blake, J.D. and Cohen, F.E. 2001. Pairwise sequence alignment below the twilight zone. J. Mol. Biol. 307 721–735. - PubMed
-
- Bowie, J.U., Reidhaar-Olson, J.F., Lim, W.A., and Sauer, R.T. 1990. Deciphering the message in protein sequences: Tolerance to amino acid substitutions. Science 247 1306–1310. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
