

A perspective on epistasis: limits of models displaying no main effect
- PMID: 11791213
- PMCID: PMC384920
- DOI: 10.1086/338759
A perspective on epistasis: limits of models displaying no main effect
Abstract
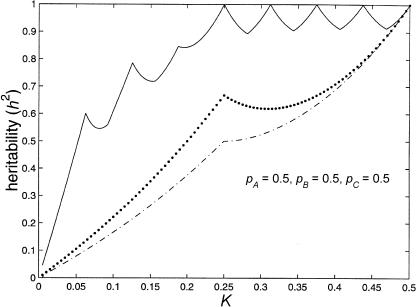
The completion of a draft sequence of the human genome and the promise of rapid single-nucleotide-polymorphism-genotyping technologies have resulted in a call for the abandonment of linkage studies in favor of genome scans for association. However, there exists a large class of genetic models for which this approach will fail: purely epistatic models with no additive or dominance variation at any of the susceptibility loci. As a result, traditional association methods (such as case/control, measured genotype, and transmission/disequilibrium test [TDT]) will have no power if the loci are examined individually. In this article, we examine this class of models, delimiting the range of genetic determination and recurrence risks for two-, three-, and four-locus purely epistatic models. Our study reveals that these models, although giving rise to no additive or dominance variation, do give rise to increased allele sharing between affected sibs. Thus, a genome scan for linkage could detect genomic subregions harboring susceptibility loci. We also discuss some simple multilocus extensions of single-locus analysis methods, including a conditional form of the TDT.
Figures

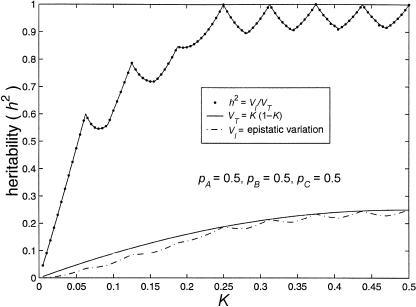
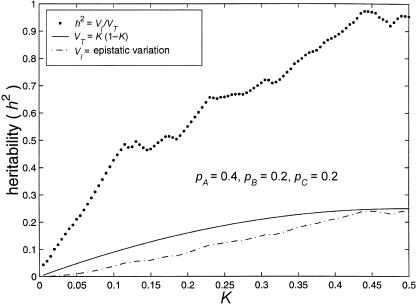
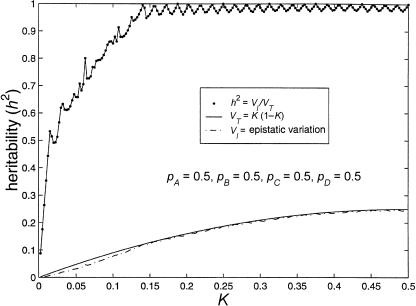
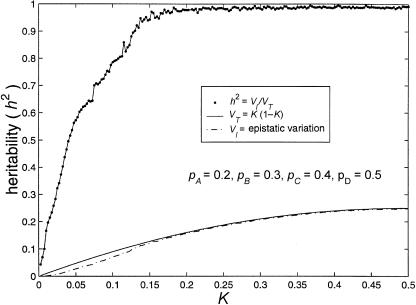
References
Electronic-Database Information
-
- “cdd and cddplus Homepage,” http://www.ifor.math.ethz.ch/~fukuda/cdd_home/cdd.html
References
-
- Boerwinkle E, Chakraborty R, Sing CF (1986) The use of measured genotype information in the analysis of quantitative phenotypes in man. I. Models and analytical methods. Ann Hum Genet 50:181–194 - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
