

Complete genome sequence of Clostridium perfringens, an anaerobic flesh-eater
- PMID: 11792842
- PMCID: PMC117419
- DOI: 10.1073/pnas.022493799
Complete genome sequence of Clostridium perfringens, an anaerobic flesh-eater
Abstract
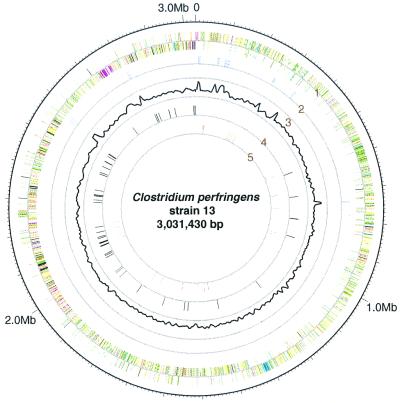
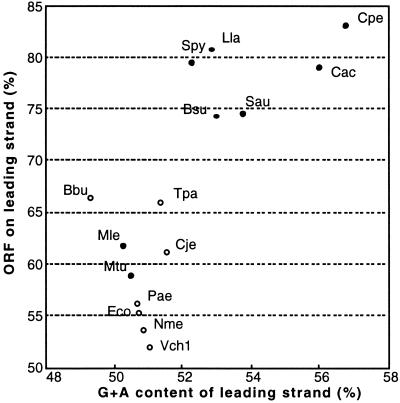
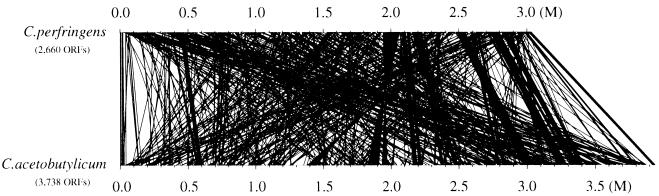
Clostridium perfringens is a Gram-positive anaerobic spore-forming bacterium that causes life-threatening gas gangrene and mild enterotoxaemia in humans, although it colonizes as normal intestinal flora of humans and animals. The organism is known to produce a variety of toxins and enzymes that are responsible for the severe myonecrotic lesions. Here we report the complete 3,031,430-bp sequence of C. perfringens strain 13 that comprises 2,660 protein coding regions and 10 rRNA genes, showing pronounced low overall G + C content (28.6%). The genome contains typical anaerobic fermentation enzymes leading to gas production but no enzymes for the tricarboxylic acid cycle or respiratory chain. Various saccharolytic enzymes were found, but many enzymes for amino acid biosynthesis were lacking in the genome. Twenty genes were newly identified as putative virulence factors of C. perfringens, and we found a total of five hyaluronidase genes that will also contribute to virulence. The genome analysis also proved an efficient method for finding four members of the two-component VirR/VirS regulon that coordinately regulates the pathogenicity of C. perfringens. Clearly, C. perfringens obtains various essential materials from the host by producing several degradative enzymes and toxins, resulting in massive destruction of the host tissues.
Figures

References
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
