

Activation and inhibition of G protein-coupled receptors by cell-penetrating membrane-tethered peptides
- PMID: 11805322
- PMCID: PMC117359
- DOI: 10.1073/pnas.022460899
Activation and inhibition of G protein-coupled receptors by cell-penetrating membrane-tethered peptides
Abstract
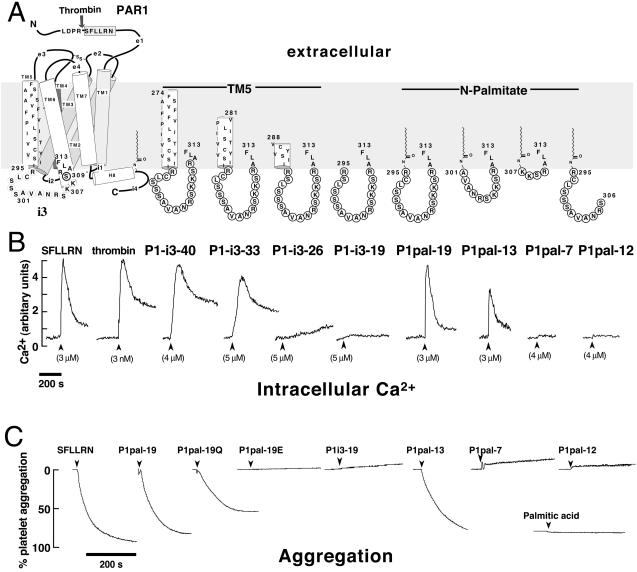
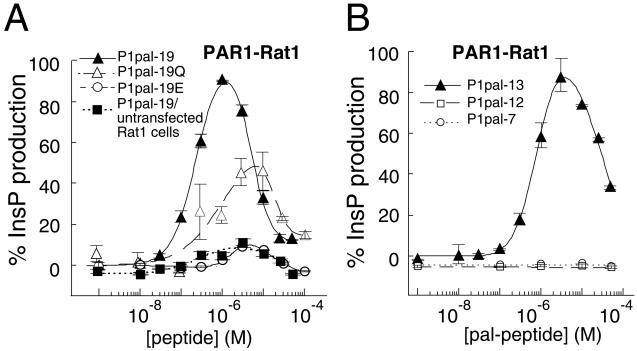
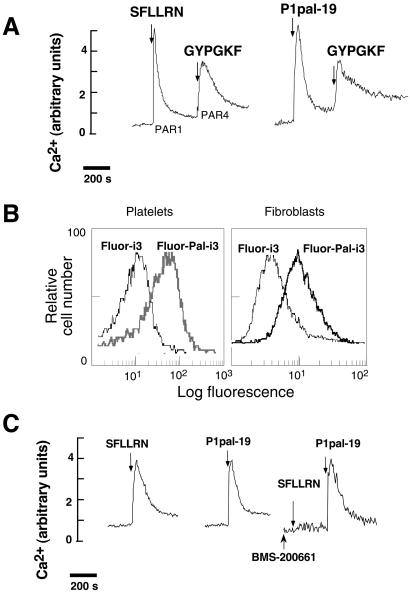
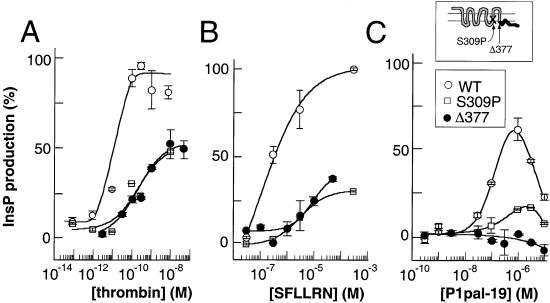
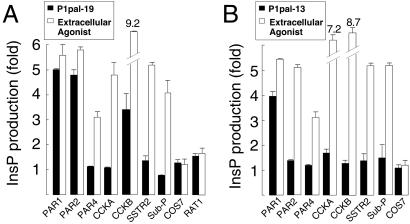
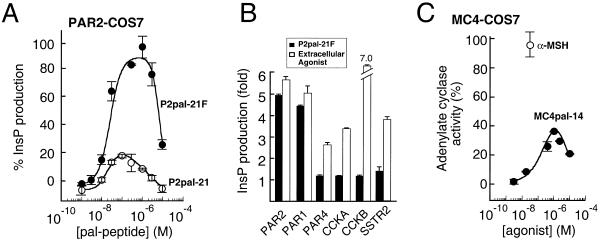
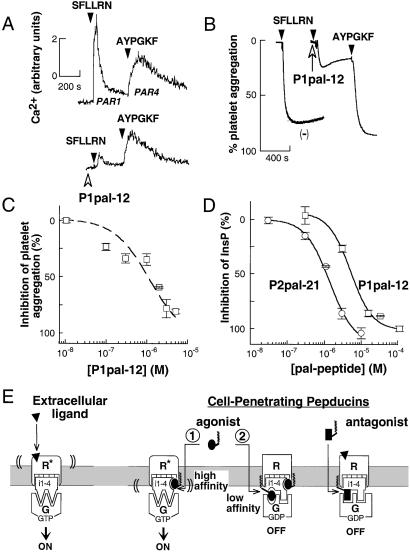
Classical ligands bind to the extracellular surface of their cognate receptors and activate signaling pathways without crossing the plasma membrane barrier. We selectively targeted the intracellular receptor-G protein interface by using cell-penetrating membrane-tethered peptides. Attachment of a palmitate group to peptides derived from the third intracellular loop of protease-activated receptors-1 and -2 and melanocortin-4 receptors yields agonists and/or antagonists of receptor-G protein signaling. These lipidated peptides--which we have termed pepducins--require the presence of their cognate receptor for activity and are highly selective for receptor type. Mutational analysis of both intact receptor and pepducins demonstrates that the cell-penetrating agonists do not activate G proteins by the same mechanism as the intact receptor third intracellular loop but instead require the C-tail of the receptor. Construction of such peptide-lipid conjugates constitutes a new molecular strategy for the development of therapeutics targeted to the receptor-effector interface.
Figures
References
-
- Palczewski K, Kumasaka T, Hori T, Behnke C A, Motoshima H, Fox B A, Le Trong I, Teller D C, Okada T, Stenkamp R E, et al. Science. 2000;289:739–745. - PubMed
-
- Gether U, Kolbilka B K. J Biol Chem. 1998;273:17979–17982. - PubMed
-
- Cotecchia S, Ostrowski J, Kjelsberg M A, Caron M G, Lefkowitz R J. J Biol Chem. 1992;267:1633–1639. - PubMed
-
- Kostenis E, Conklin B R, Wess J. Biochemistry. 1997;36:1487–1495. - PubMed
-
- Kjelsberg M A, Cotecchia S, Ostrowski J, Caron M G, Lefkowitz R J. J Biol Chem. 1992;267:1430–1433. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
