

Cell proliferation and DNA breaks are involved in ultraviolet light-induced apoptosis in nucleotide excision repair-deficient Chinese hamster cells
- PMID: 11809844
- PMCID: PMC65093
- DOI: 10.1091/mbc.01-05-0225
Cell proliferation and DNA breaks are involved in ultraviolet light-induced apoptosis in nucleotide excision repair-deficient Chinese hamster cells
Abstract
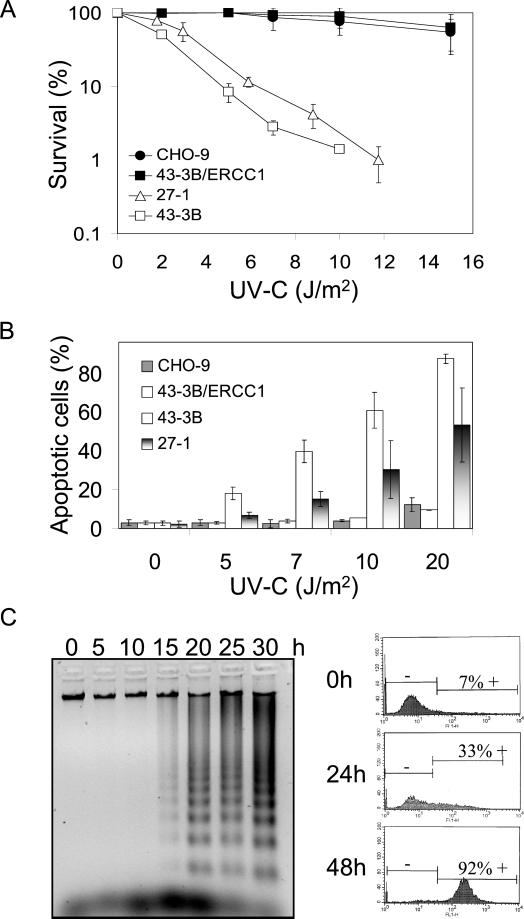
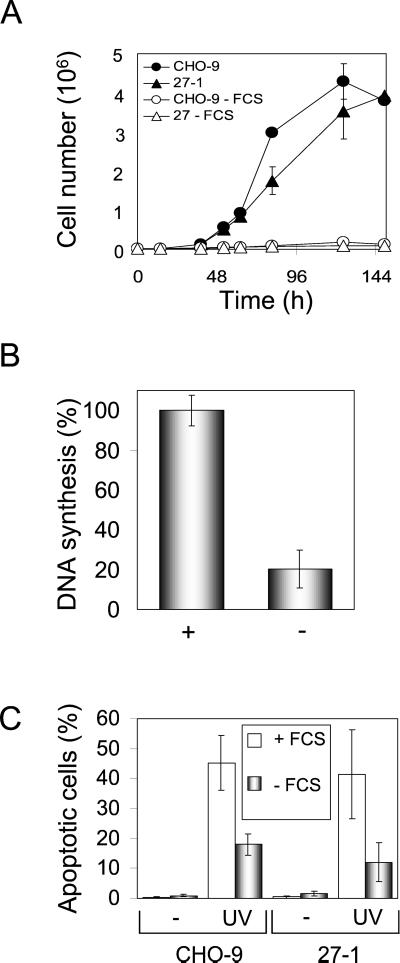
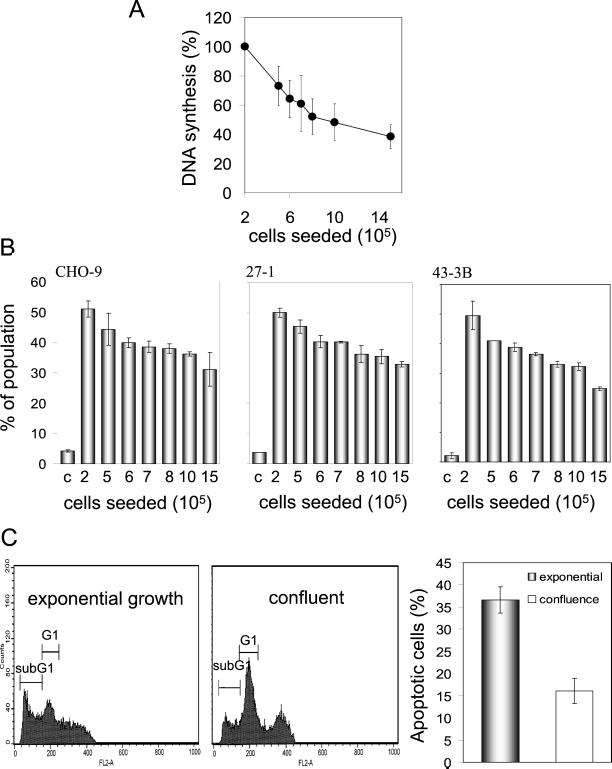
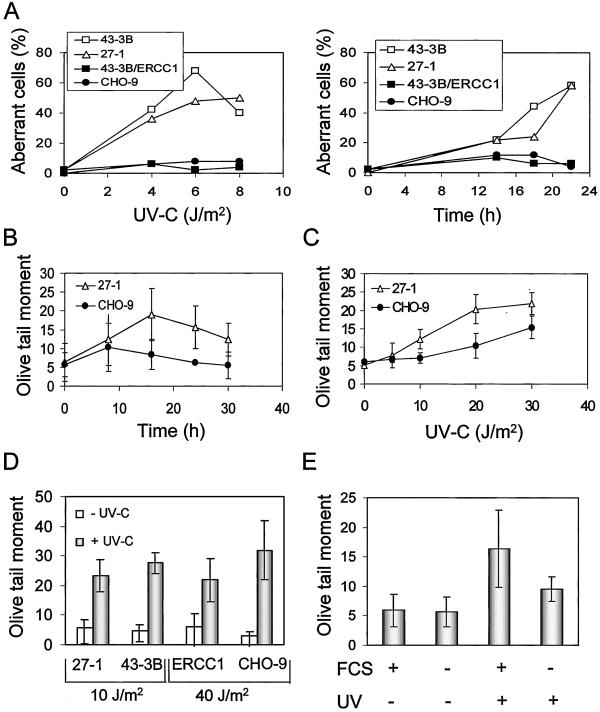
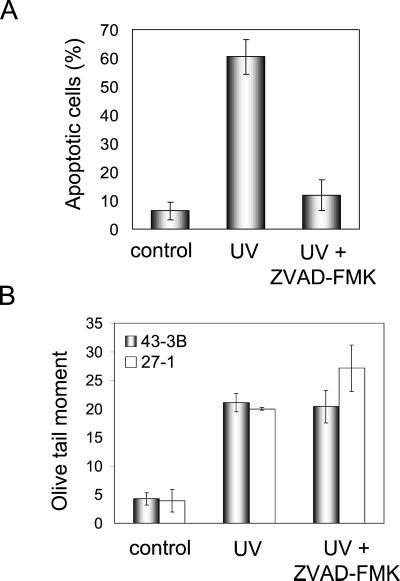
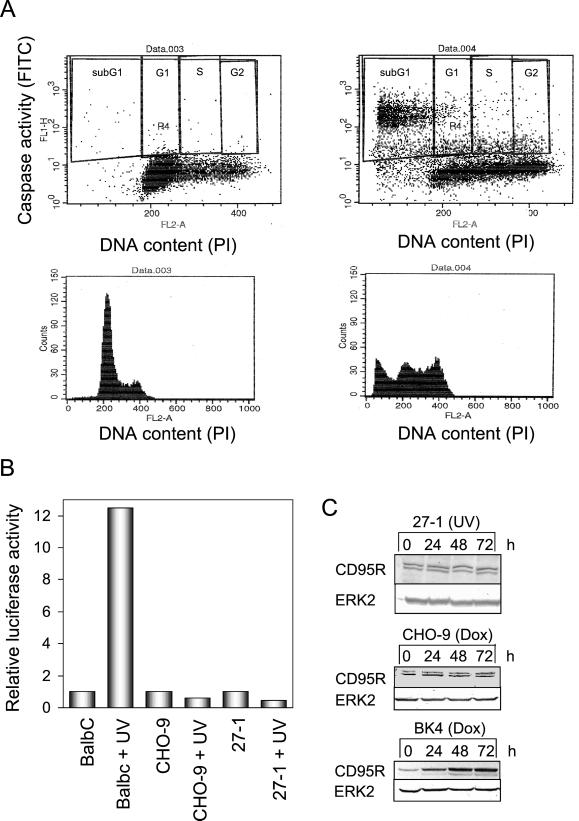
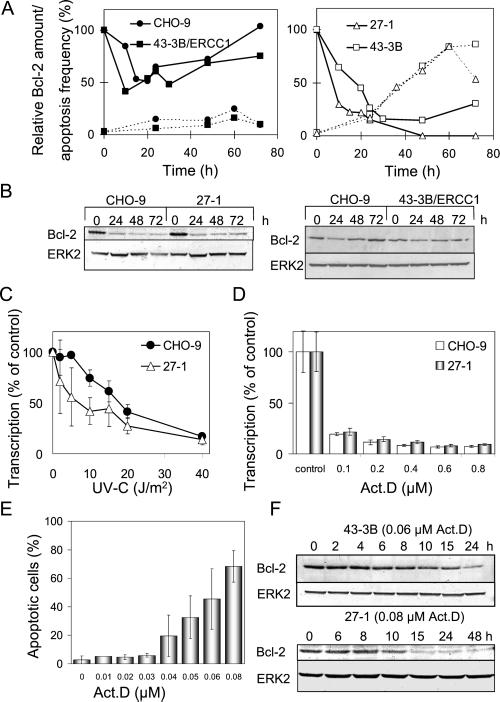
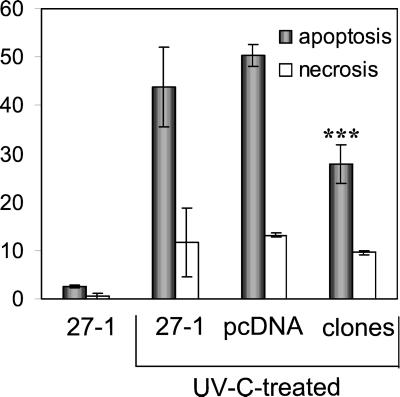
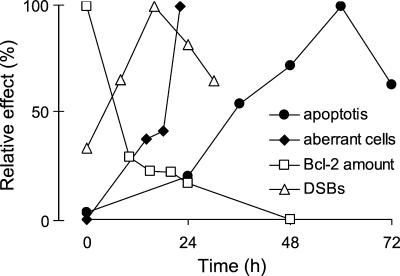
UV light targets both membrane receptors and nuclear DNA, thus evoking signals triggering apoptosis. Although receptor-mediated apoptosis has been extensively investigated, the role of DNA damage in apoptosis is less clear. To analyze the importance of DNA damage induced by UV-C light in apoptosis, we compared nucleotide excision repair (NER)-deficient Chinese hamster ovary cells (lines 27-1 and 43-3B mutated for the repair genes ERCC3 and ERCC1, respectively) with the corresponding DNA repair-proficient fibroblasts (CHO-9 and ERCC1 complemented 43-3B cells). NER-deficient cells were hypersensitive as to the induction of apoptosis, indicating that apoptosis induced by UV-C light is due to unrepaired DNA base damage. Unrepaired lesions, however, do not activate the apoptotic pathway directly because apoptosis upon UV-C irradiation requires DNA replication and cell proliferation. It is also shown that in NER-deficient cells unrepaired lesions are converted into DNA double-strand breaks (DSBs) and chromosomal aberrations by a replication-dependent process that precedes apoptosis. We therefore propose that DSBs arising from replication of DNA containing nonrepaired lesions act as an ultimate trigger of UV-C-induced apoptosis. Induction of apoptosis by UV-C light was related to decline in the expression level of Bcl-2 and activation of caspases. Decline of Bcl-2 and subsequent apoptosis might also be caused, at least in part, by UV-C-induced blockage of transcription, which was more pronounced in NER-deficient than in wild-type cells. This is in line with experiments with actinomycin D, which provoked Bcl-2 decline and apoptosis. UV-C-induced apoptosis due to nonrepaired DNA lesions, replication-dependent formation of DSBs, and activation of the mitochondrial damage pathway is independent of functional p53 for which the cells are mutated.
Figures
References
-
- Antonsson B, Martinou JC. The Bcl-2 protein family. Exp Cell Res. 2000;256:50–57. - PubMed
-
- Brash DE, Ziegler A, Jonason AS, Simon JA, Kunala S, Leffell DJ. Sunlight and sunburn in human skin cancer: p53, apoptosis, and tumor promotion. J Invest Dermatol Symp Proc. 1996;2:136–142. - PubMed
-
- Canman CE, Kastan MB. Signal transduction. Three paths to stress relief. Nature. 1996;384:213–214. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
