

Constitutively active AMP kinase mutations cause glycogen storage disease mimicking hypertrophic cardiomyopathy
- PMID: 11827995
- PMCID: PMC150860
- DOI: 10.1172/JCI14571
Constitutively active AMP kinase mutations cause glycogen storage disease mimicking hypertrophic cardiomyopathy
Abstract
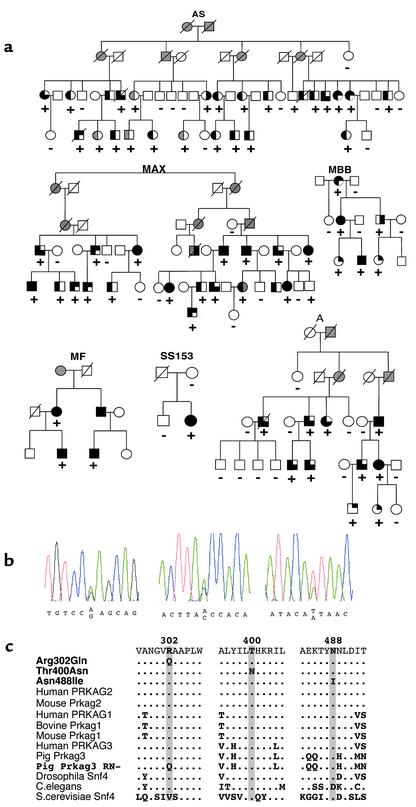
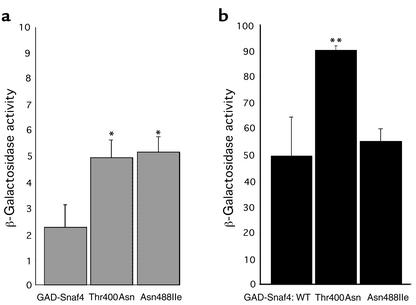
Mutations in PRKAG2, the gene for the gamma 2 regulatory subunit of AMP-activated protein kinase, cause cardiac hypertrophy and electrophysiologic abnormalities, particularly preexcitation (Wolff-Parkinson-White syndrome) and atrioventricular conduction block. To understand the mechanisms by which PRKAG2 defects cause disease, we defined novel mutations, characterized the associated cardiac histopathology, and studied the consequences of introducing these mutations into the yeast homologue of PRKAG2, Snf4. Although the cardiac pathology caused by PRKAG2 mutations Arg302Gln, Thr400Asn, and Asn488Ile include myocyte enlargement and minimal interstitial fibrosis, these mutations were not associated with myocyte and myofibrillar disarray, the pathognomonic features of hypertrophic cardiomyopathy caused by sarcomere protein mutations. Instead PRKAG2 mutations caused pronounced vacuole formation within myocytes. Several lines of evidence indicated these vacuoles were filled with glycogen-associated granules. Analyses of the effects of human PRKAG2 mutations on Snf1/Snf4 kinase function demonstrated constitutive activity, which could foster glycogen accumulation. Taken together, our data indicate that PRKAG2 mutations do not cause hypertrophic cardiomyopathy but rather lead to a novel myocardial metabolic storage disease, in which hypertrophy, ventricular pre-excitation and conduction system defects coexist.
Figures

References
-
- Kemp BE, et al. Dealing with energy demand: the AMP-activated protein kinase. Trends Biochem Sci. 1999;24:22–25. - PubMed
-
- Blair E, et al. Mutations in the gamma(2) subunit of AMP-activated protein kinase cause familial hypertrophic cardiomyopathy: evidence for the central role of energy compromise in disease pathogenesis. Hum Mol Genet. 2001;10:1215–1220. - PubMed
-
- Gollob MH, et al. Identification of a gene responsible for familial Wolff-Parkinson-White syndrome. N Engl J Med. 2001;344:1823–1831. - PubMed
-
- Fatkin, D., Seidman, J.G., and Seidman, C.E. 2000. Hypertrophic cardiomyopathy. In Cardiovascular medicine. J.T. Willerson and J.N. Cohn, editors. W.B. Saunders Co. Philadelphia, Pennsylvania, USA. 1055–1074.
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
