

Degradation of STAT1 and STAT2 by the V proteins of simian virus 5 and human parainfluenza virus type 2, respectively: consequences for virus replication in the presence of alpha/beta and gamma interferons
- PMID: 11836393
- PMCID: PMC153821
- DOI: 10.1128/jvi.76.5.2159-2167.2002
Degradation of STAT1 and STAT2 by the V proteins of simian virus 5 and human parainfluenza virus type 2, respectively: consequences for virus replication in the presence of alpha/beta and gamma interferons
Abstract
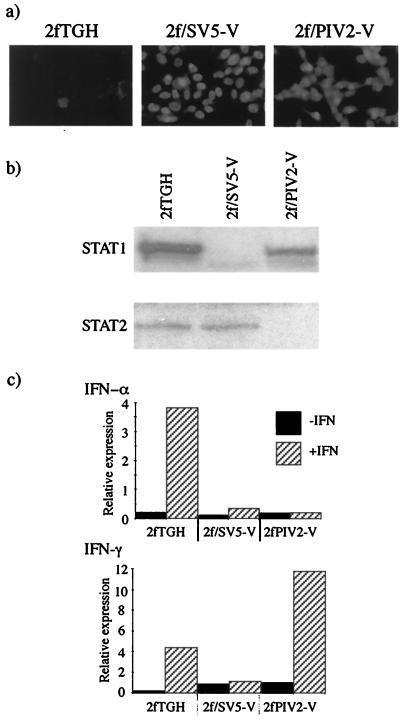
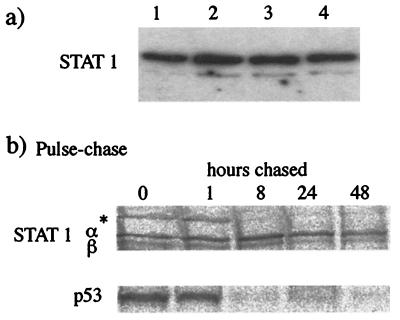
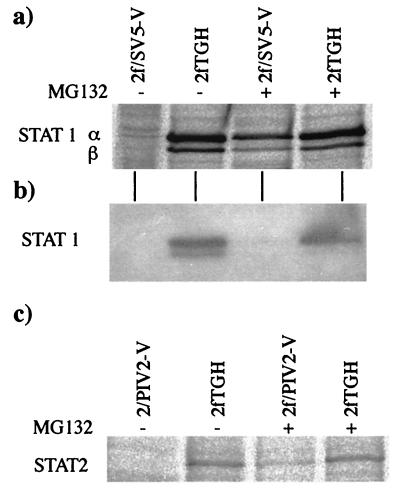
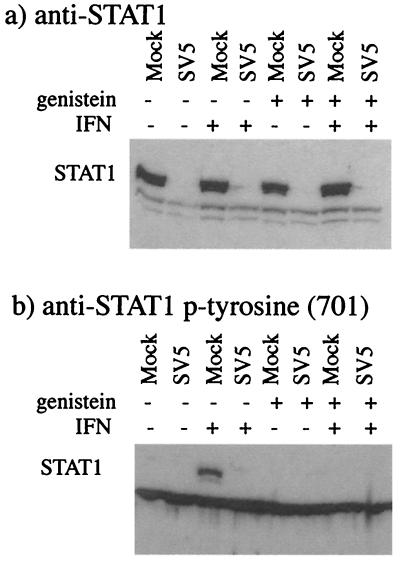
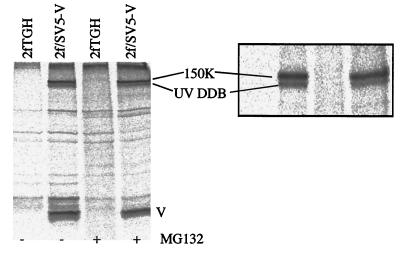
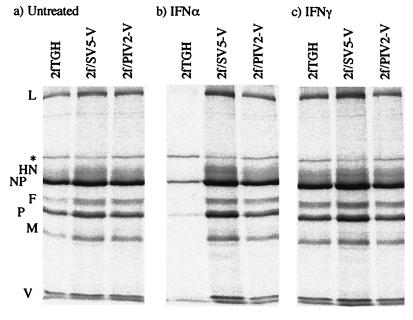
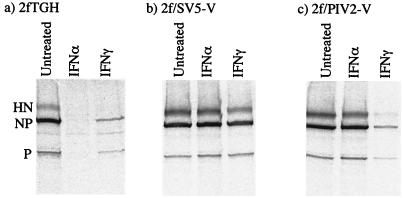
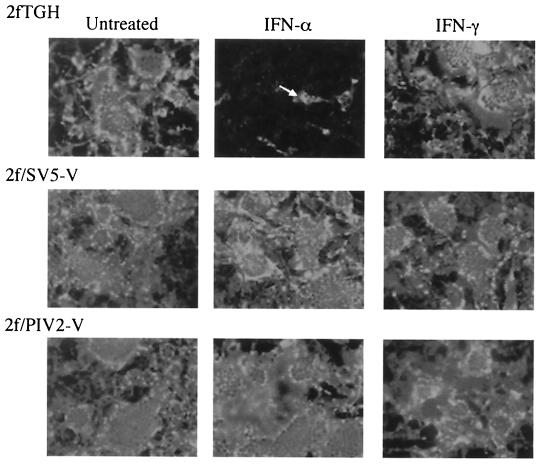
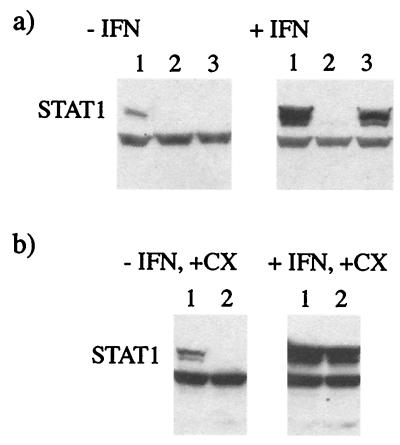
Human cell lines were isolated that express the V protein of either simian virus 5 (SV5) or human parainfluenza virus type 2 (hPIV2); the cell lines were termed 2f/SV5-V and 2f/PIV2-V, respectively. STAT1 was not detectable in 2f/SV5-V cells, and the cells failed to signal in response to either alpha/beta interferons (IFN-alpha and IFN-beta, or IFN-alpha/beta) or gamma interferon (IFN-gamma). In contrast, STAT2 was absent from 2f/PIV2-V cells, and IFN-alpha/beta but not IFN-gamma signaling was blocked in these cells. Treatment of both 2f/SV5-V and 2f/PIV2-V cells with a proteasome inhibitor allowed the respective STAT levels to accumulate at rates similar to those seen in 2fTGH cells, indicating that the V proteins target the STATs for proteasomal degradation. Infection with SV5 can lead to a complete loss of both phosphorylated and nonphosphorylated forms of STAT1 by 6 h postinfection. Since the turnover of STAT1 in uninfected cells is longer than 24 h, we conclude that degradation of STAT1 is the main mechanism by which SV5 blocks interferon (IFN) signaling. Pretreatment of 2fTGH cells with IFN-alpha severely inhibited both SV5 and hPIV2 protein synthesis. However, and in marked contrast, pretreatment of 2fTGH cells with IFN-gamma had little obvious effect on SV5 protein synthesis but did significantly reduce the replication of hPIV2. Pretreament with IFN-alpha or IFN-gamma did not induce an antiviral state in 2f/SV5-V cells, indicating either that the induction of an antiviral state is completely dependent on STAT signaling or that the V protein interferes with other, STAT-independent cell signaling pathways that may be induced by IFNs. Even though SV5 blocked IFN signaling, the addition of exogenous IFN-alpha to the culture medium of 2fTGH cells 12 h after a low-multiplicity infection with SV5 significantly reduced the subsequent cell-to-cell spread of virus. The significance of the results in terms of the strategy that these viruses have evolved to circumvent the IFN response is discussed.
Figures
References
-
- Choppin, P. W. 1964. Multiplication of a myxovirus (SV5) with minimal cytopathic effects and without interference. Virology 23:224-233. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
