

Single-domain antibody fragments with high conformational stability
- PMID: 11847273
- PMCID: PMC2373476
- DOI: 10.1110/ps.34602
Single-domain antibody fragments with high conformational stability
Abstract
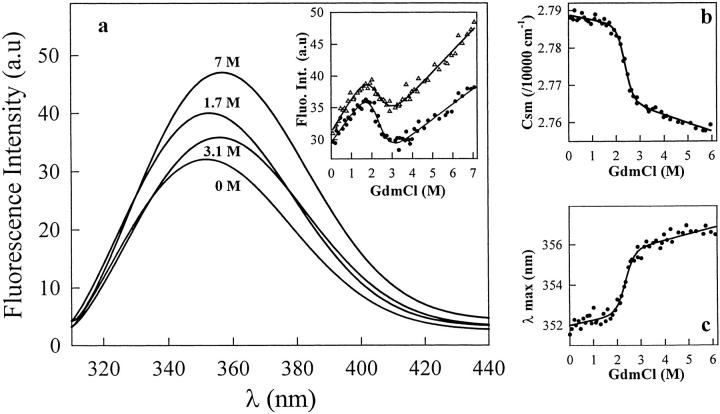
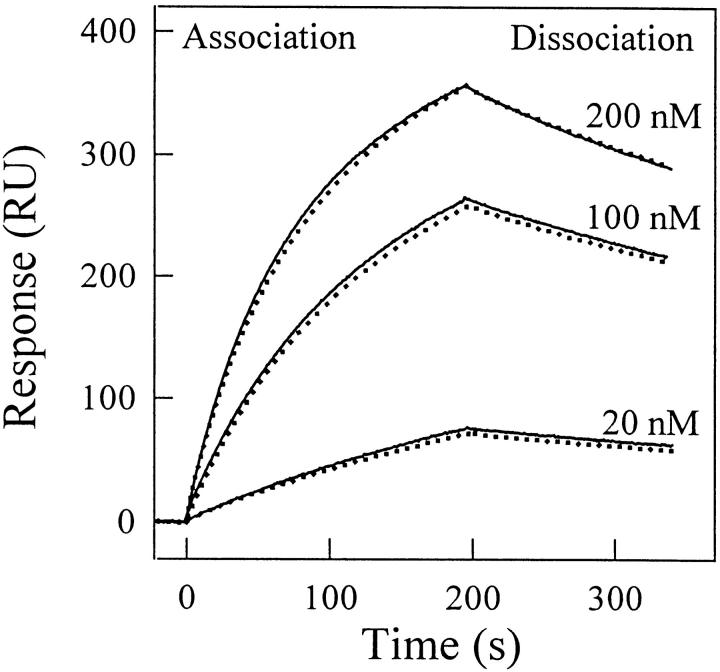
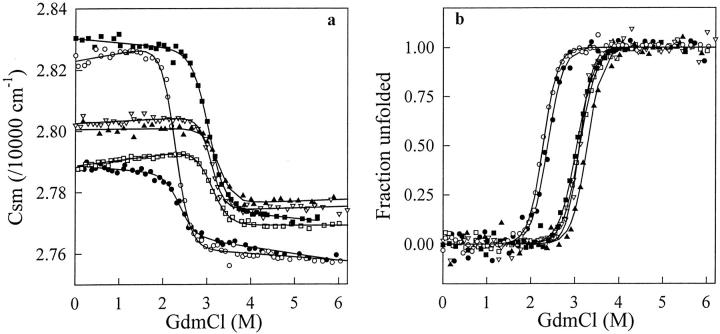
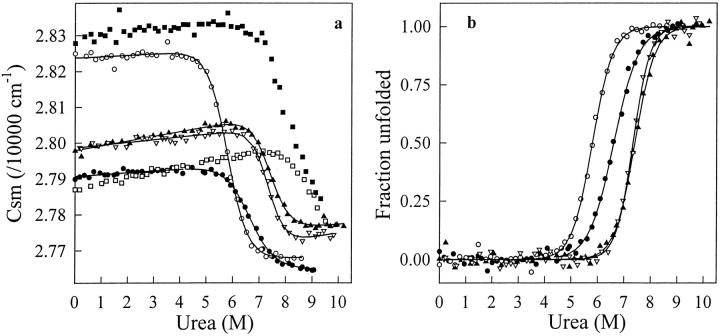
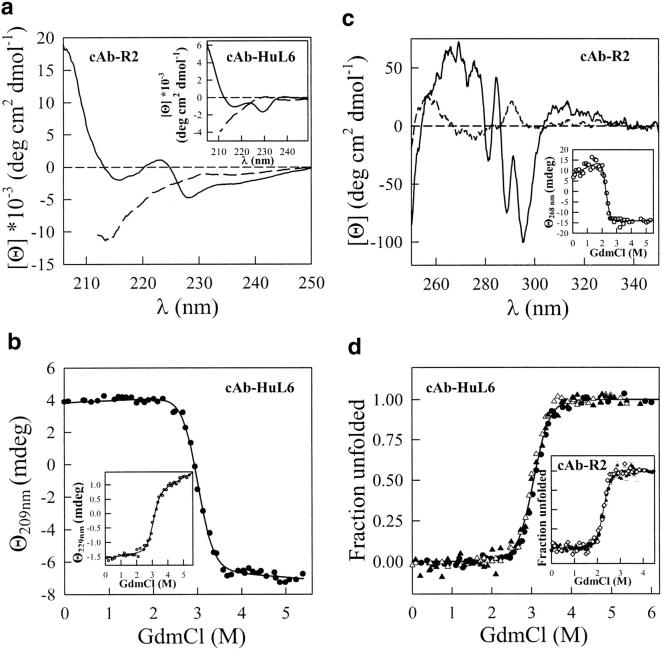
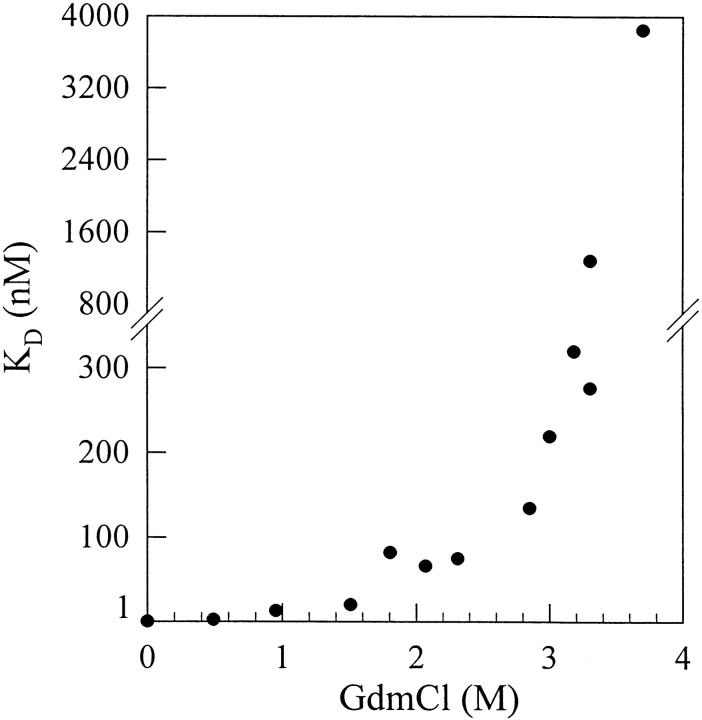
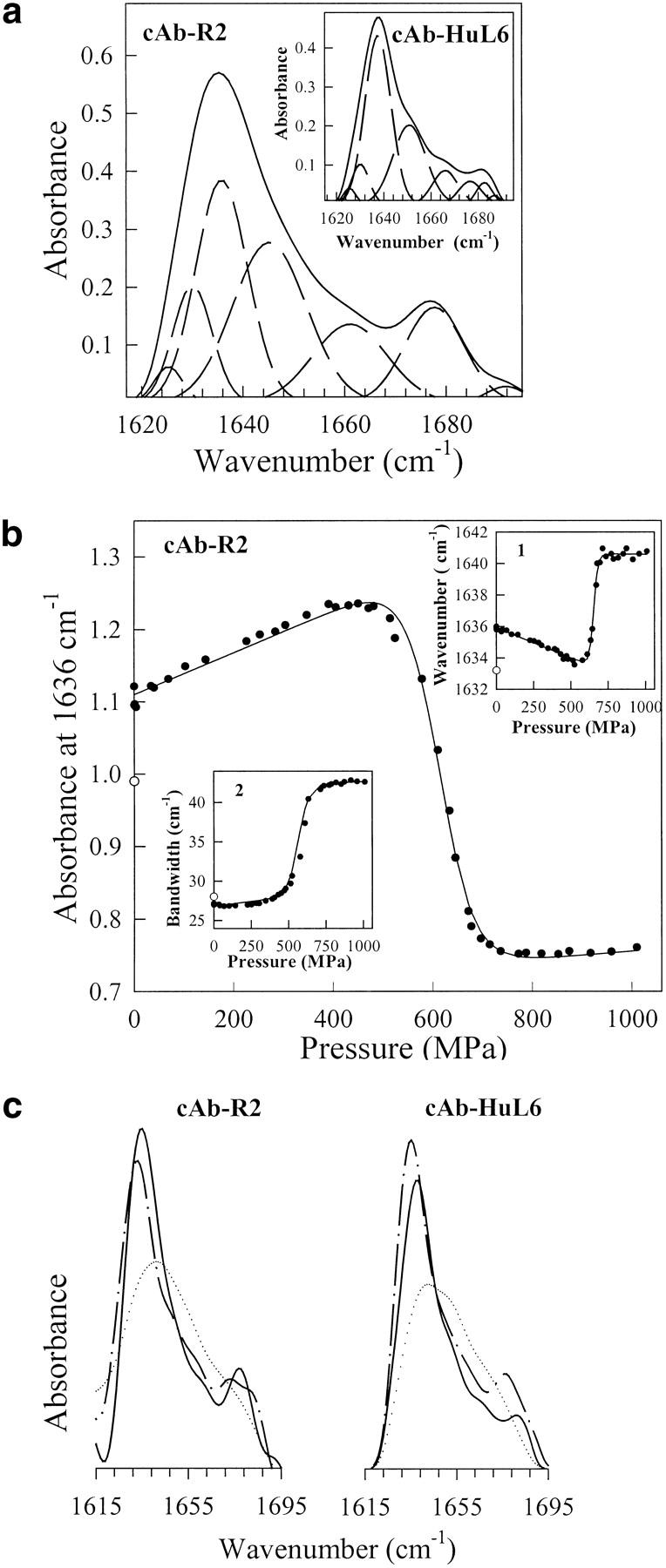
A variety of techniques, including high-pressure unfolding monitored by Fourier transform infrared spectroscopy, fluorescence, circular dichroism, and surface plasmon resonance spectroscopy, have been used to investigate the equilibrium folding properties of six single-domain antigen binders derived from camelid heavy-chain antibodies with specificities for lysozymes, beta-lactamases, and a dye (RR6). Various denaturing conditions (guanidinium chloride, urea, temperature, and pressure) provided complementary and independent methods for characterizing the stability and unfolding properties of the antibody fragments. With all binders, complete recovery of the biological activity after renaturation demonstrates that chemical-induced unfolding is fully reversible. Furthermore, denaturation experiments followed by optical spectroscopic methods and affinity measurements indicate that the antibody fragments are unfolded cooperatively in a single transition. Thus, unfolding/refolding equilibrium proceeds via a simple two-state mechanism (N <--> U), where only the native and the denatured states are significantly populated. Thermally-induced denaturation, however, is not completely reversible, and the partial loss of binding capacity might be due, at least in part, to incorrect refolding of the long loops (CDRs), which are responsible for antigen recognition. Most interestingly, all the fragments are rather resistant to heat-induced denaturation (apparent T(m) = 60-80 degrees C), and display high conformational stabilities (DeltaG(H(2)O) = 30-60 kJ mole(-1)). Such high thermodynamic stability has never been reported for any functional conventional antibody fragment, even when engineered antigen binders are considered. Hence, the reduced size, improved solubility, and higher stability of the camelid heavy-chain antibody fragments are of special interest for biotechnological and medical applications.
Figures

References
-
- Artymiuk, P.J. and Blake, C.C.F. 1981. Refinement of human lysozyme at 1.5 Å resolution. J. Mol. Biol. 152: 737–762. - PubMed
-
- Better, M., Chang, C.P., Robinson, R.R., and Horwitz, A.H. 1988. Escherichia coli secretion of an active chimeric antibody fragment. Science 240: 1041–1043. - PubMed
-
- Bird, R.E., Hardman, K.D., Jacobson, J.W., Johnson, S., Kaufman, B.M., Lee, S.M., Lee, T., Pope, S.H., Riordan, G.S., and Whitlow, M. 1988. Single-chain antigen-binding proteins. Science 242: 423–426. - PubMed
-
- Blake, C.C.F., Koenig, D.F., Mair, G.A., and Sarma, R. 1965. Crystal structure of lysozyme by X-ray diffraction. Nature 206: 757–761. - PubMed
-
- Byler, D.M. and Susi., H. 1986. Examination of the secondary structure of proteins by deconvolved FTIR spectra. Biopolymers 25: 469–487. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
