

Ionic mechanism of ouabain-induced concurrent apoptosis and necrosis in individual cultured cortical neurons
- PMID: 11850462
- PMCID: PMC6757565
- DOI: 10.1523/JNEUROSCI.22-04-01350.2002
Ionic mechanism of ouabain-induced concurrent apoptosis and necrosis in individual cultured cortical neurons
Abstract
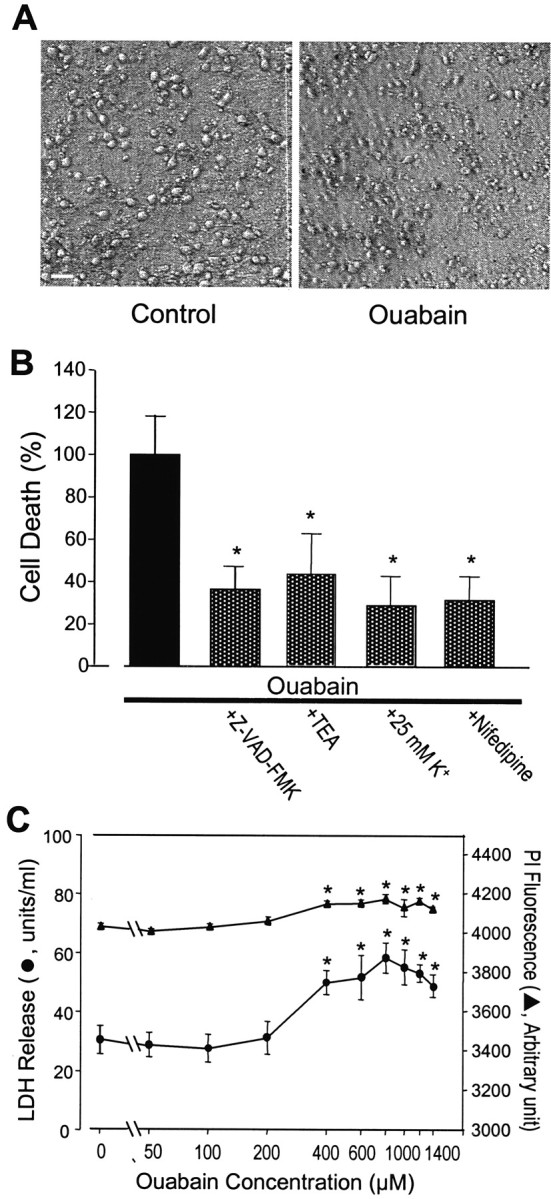
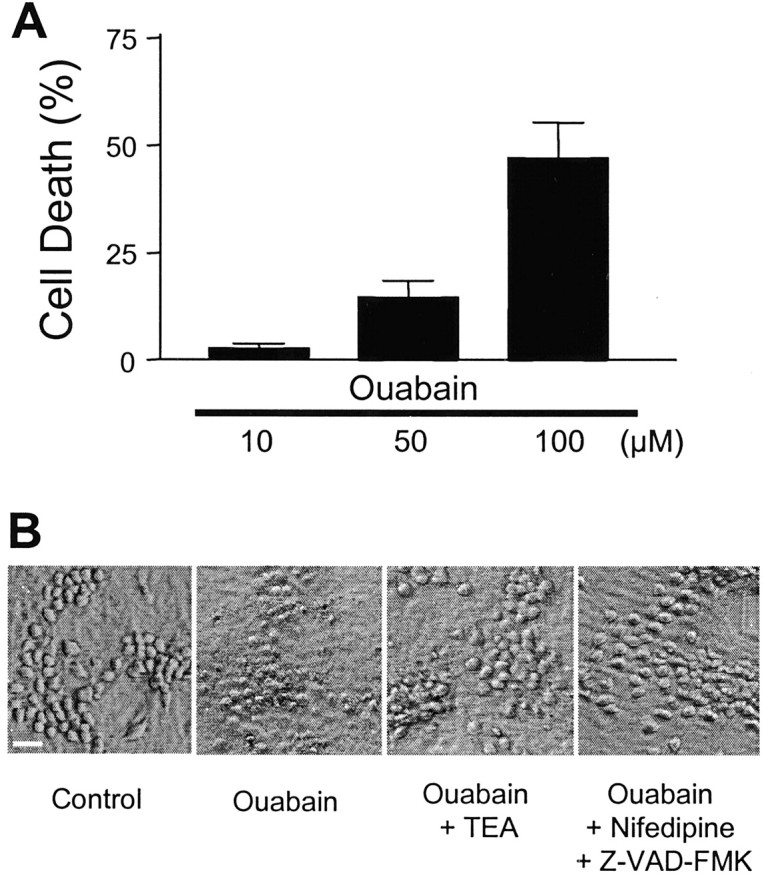
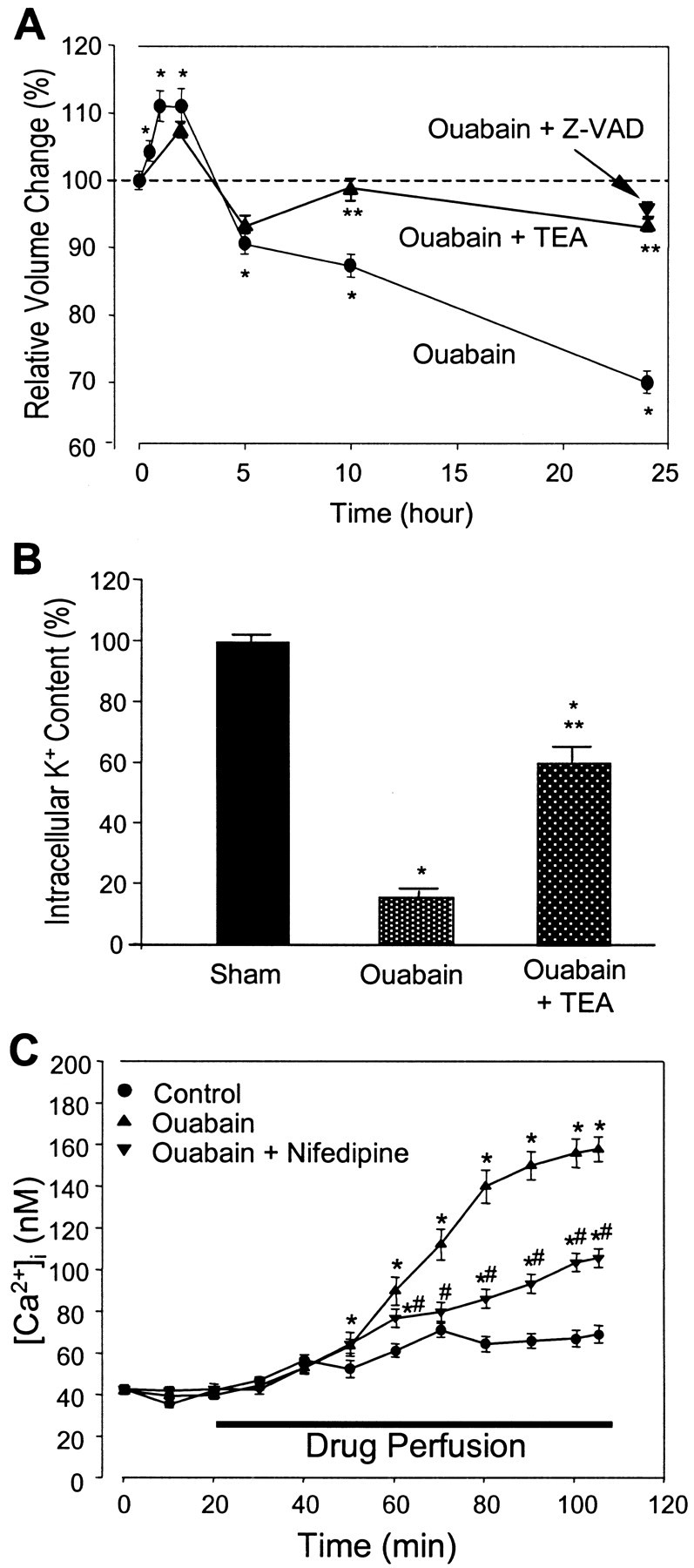
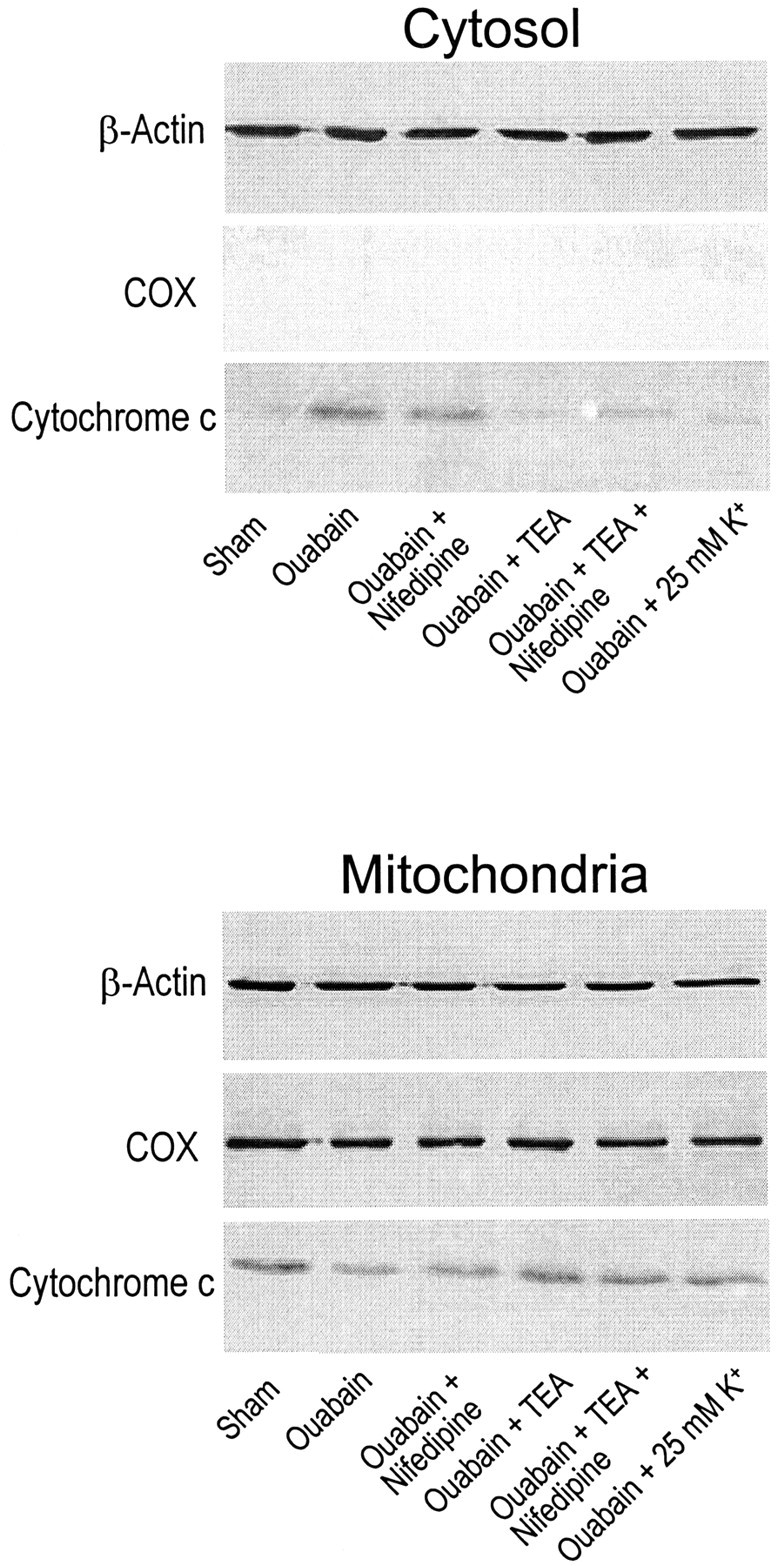
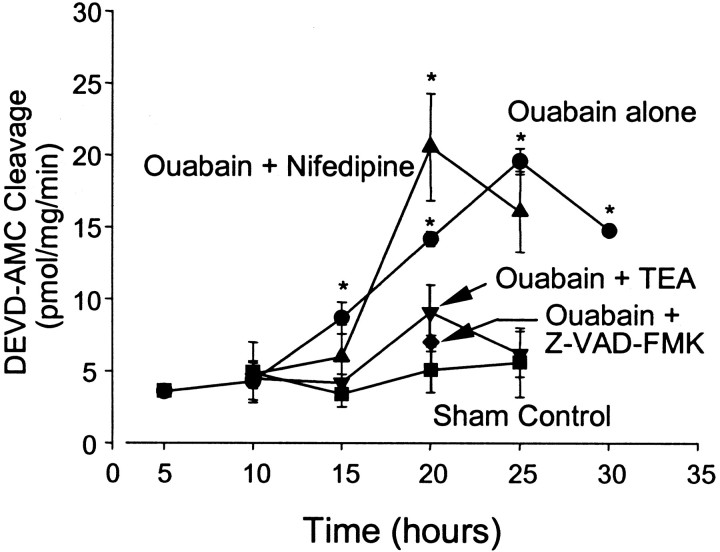
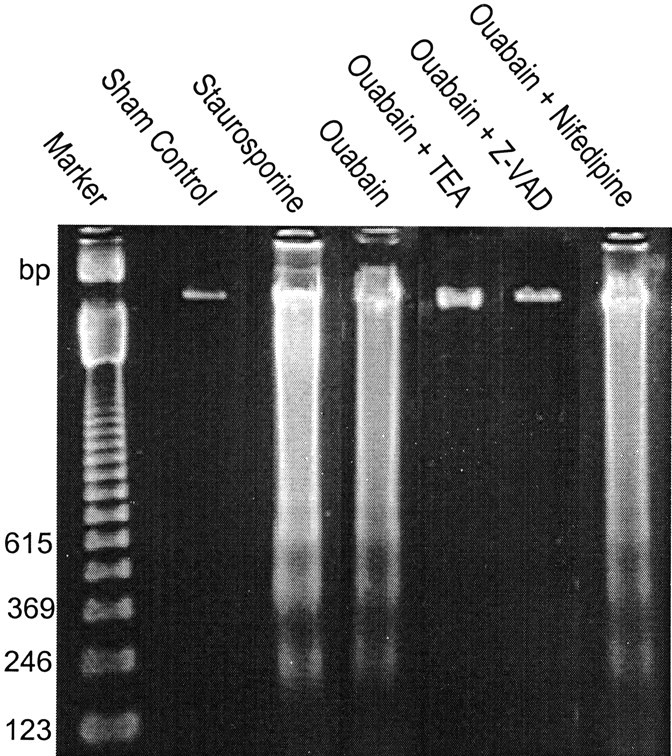
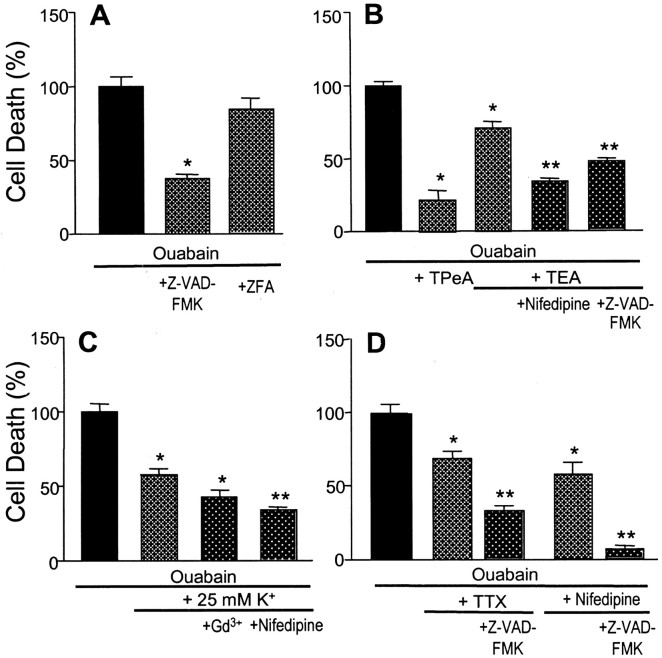
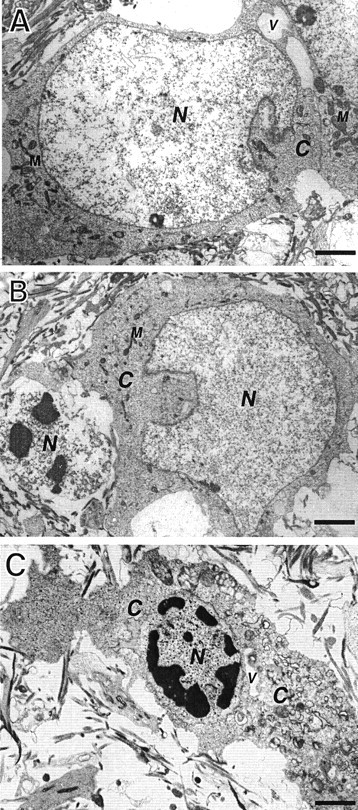
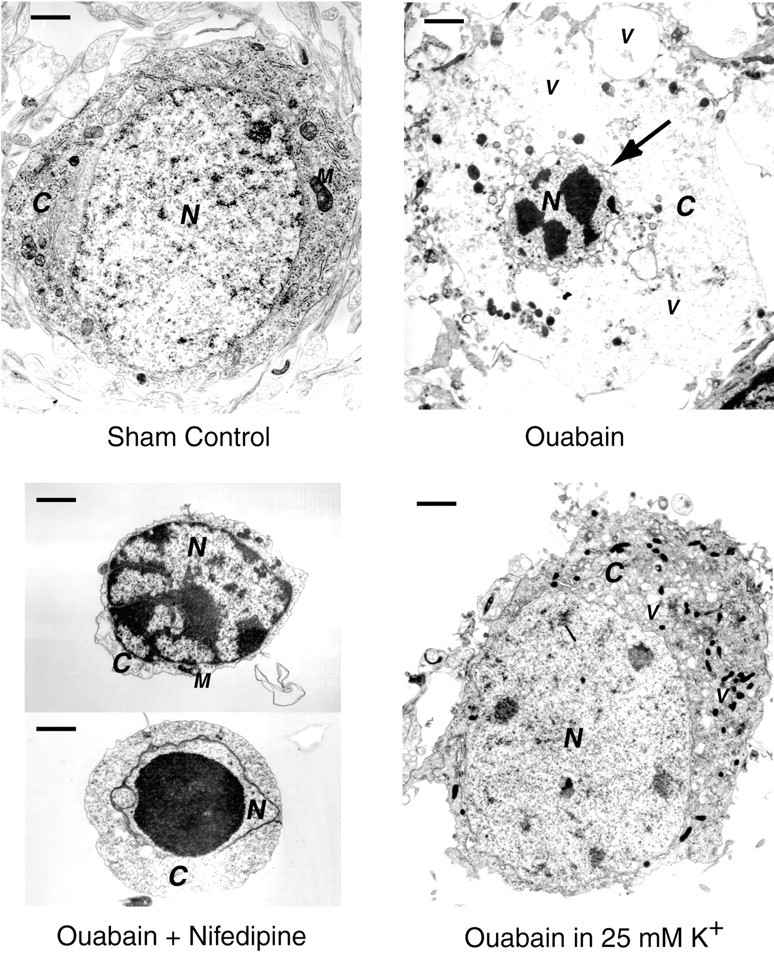
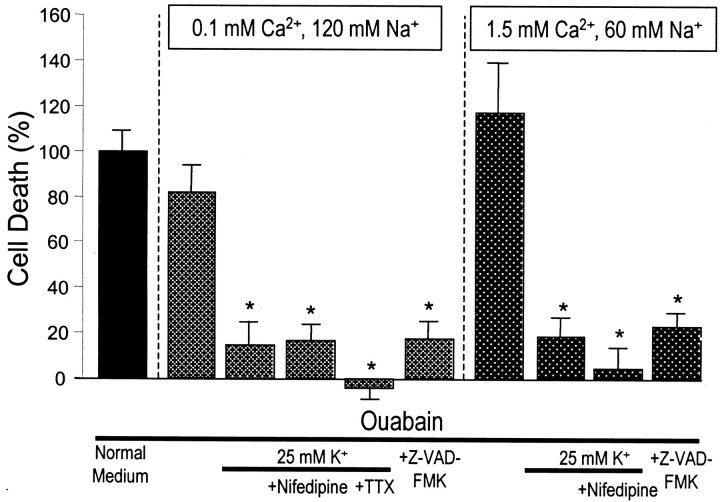
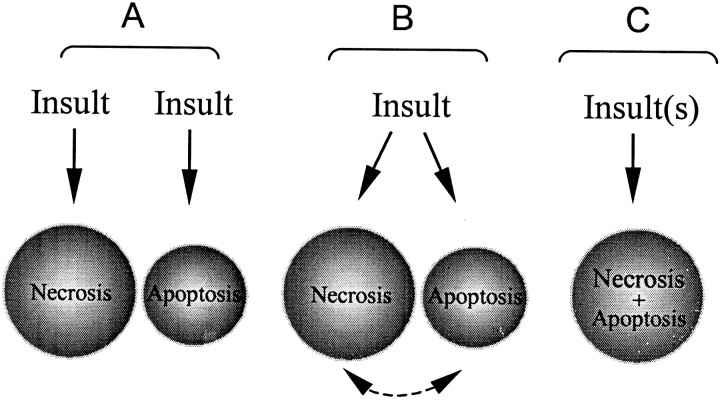
Energy deficiency and dysfunction of the Na+, K+-ATPase are common consequences of many pathological insults. The nature and mechanism of cell injury induced by impaired Na+, K+-ATPase, however, are not well defined. We used cultured cortical neurons to examine the hypothesis that blocking the Na+, K+-ATPase induces apoptosis by depleting cellular K+ and, concurrently, induces necrotic injury in the same cells by increasing intracellular Ca2+ and Na+. The Na+, K+-ATPase inhibitor ouabain induced concentration-dependent neuronal death. Ouabain triggered transient neuronal cell swelling followed by cell shrinkage, accompanied by intracellular Ca2+ and Na+ increase, K+ decrease, cytochrome c release, caspase-3 activation, and DNA laddering. Electron microscopy revealed the coexistence of ultrastructural features of both apoptosis and necrosis in individual cells. The caspase inhibitor Z-Val-Ala-Asp(OMe)-fluoromethyl ketone (Z-VAD-FMK) blocked >50% of ouabain-induced neuronal death. Potassium channel blockers or high K+ medium, but not Ca2+ channel blockade, prevented cytochrome c release, caspase activation, and DNA damage. Blocking of K+, Ca2+, or Na+ channels or high K+ medium each attenuated the ouabain-induced cell death; combined inhibition of K+ channels and Ca2+ or Na+ channels resulted in additional protection. Moreover, coapplication of Z-VAD-FMK and nifedipine produced virtually complete neuroprotection. These results suggest that the neuronal death associated with Na+, K+-pump failure consists of concurrent apoptotic and necrotic components, mediated by intracellular depletion of K+ and accumulation of Ca2+ and Na+, respectively. The ouabain-induced hybrid death may represent a distinct form of cell death related to the brain injury of inadequate energy supply and disrupted ion homeostasis.
Figures
Similar articles
-
Slight impairment of Na+,K+-ATPase synergistically aggravates ceramide- and beta-amyloid-induced apoptosis in cortical neurons.Brain Res. 2002 Nov 15;955(1-2):253-9. doi: 10.1016/s0006-8993(02)03472-8. Brain Res. 2002. PMID: 12419544
-
Na+/K+ pump and endothelial cell survival: [Na+]i/[K+]i-independent necrosis triggered by ouabain, and protection against apoptosis mediated by elevation of [Na+]i.Pflugers Arch. 2004 Jun;448(3):335-45. doi: 10.1007/s00424-004-1262-9. Epub 2004 Apr 7. Pflugers Arch. 2004. PMID: 15069561
-
Block of Na+,K+-ATPase and induction of hybrid death by 4-aminopyridine in cultured cortical neurons.J Pharmacol Exp Ther. 2003 May;305(2):502-6. doi: 10.1124/jpet.102.045013. Epub 2003 Jan 21. J Pharmacol Exp Ther. 2003. PMID: 12606650
-
Volume changes in neurons: hyperexcitability and neuronal death.Contrib Nephrol. 2006;152:221-240. doi: 10.1159/000096326. Contrib Nephrol. 2006. PMID: 17065815 Review.
-
Na(+), K(+)-ATPase: the new face of an old player in pathogenesis and apoptotic/hybrid cell death.Biochem Pharmacol. 2003 Oct 15;66(8):1601-9. doi: 10.1016/s0006-2952(03)00531-8. Biochem Pharmacol. 2003. PMID: 14555240 Review.
Cited by
-
Effect of ouabain on calcium signaling in rodent brain: A systematic review of in vitro studies.Front Pharmacol. 2022 Aug 29;13:916312. doi: 10.3389/fphar.2022.916312. eCollection 2022. Front Pharmacol. 2022. PMID: 36105192 Free PMC article.
-
The MgtC virulence factor of Salmonella enterica serovar Typhimurium activates Na(+),K(+)-ATPase.J Bacteriol. 2006 Aug;188(15):5586-94. doi: 10.1128/JB.00296-06. J Bacteriol. 2006. PMID: 16855249 Free PMC article.
-
Monovalent ions and stress-induced senescence in human mesenchymal endometrial stem/stromal cells.Sci Rep. 2022 Jul 1;12(1):11194. doi: 10.1038/s41598-022-15490-2. Sci Rep. 2022. PMID: 35778548 Free PMC article.
-
Cardiac Glycosides in Human Physiology and Disease: Update for Entomologists.Insects. 2019 Apr 10;10(4):102. doi: 10.3390/insects10040102. Insects. 2019. PMID: 30974764 Free PMC article. Review.
-
Apelin Protects Primary Rat Retinal Pericytes from Chemical Hypoxia-Induced Apoptosis.J Ophthalmol. 2015;2015:186946. doi: 10.1155/2015/186946. Epub 2015 Sep 27. J Ophthalmol. 2015. PMID: 26491547 Free PMC article.
References
-
- Ahlemeyer B, Weintraut H, Schoner W. Cultured chick-embryo heart cells respond differently to ouabain as measured by the increase in their intracellular Na+ concentration. Biochem Biophys Acta. 1992;1137:135–142. - PubMed
-
- Ameisen JC. Programmed cell death (apoptosis) and cell survival regulation: relevance to AIDS and cancer. AIDS. 1994;8:1197–1213. - PubMed
-
- Ankarcrona M, Dypbukt JM, Bonfoco E, Zhivotovsky B, Orrenius S, Lipton SA, Nicotera P. Glutamate-induced neuronal death: a succession of necrosis or apoptosis depending on mitochondrial function. Neuron. 1995;15:961–973. - PubMed
-
- Archibald JT, White TD. Rapid reversal of internal Na+ and K+ contents of synaptosomes by ouabain. Nature. 1974;252:595–597. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous