

Review
doi: 10.1038/sj.bjp.0704548.
Role of bradykinin in preconditioning and protection of the ischaemic myocardium
Affiliations
- PMID: 11861312
- PMCID: PMC1573212
- DOI: 10.1038/sj.bjp.0704548
Item in Clipboard
Review
Role of bradykinin in preconditioning and protection of the ischaemic myocardium
Br J Pharmacol.
2002 Feb.
No abstract available
Figures
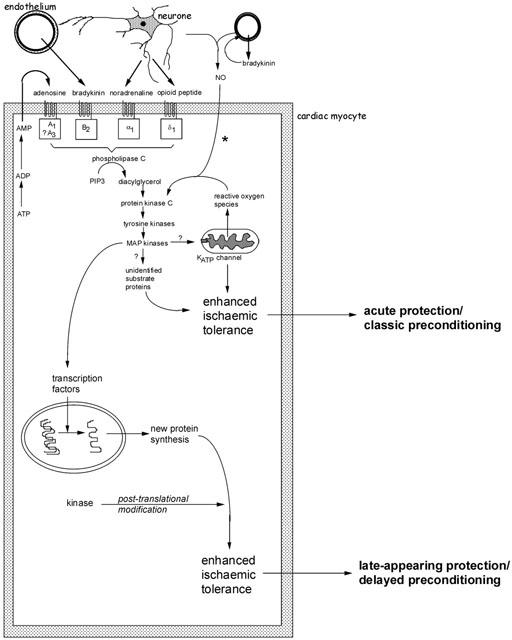
Schematic representation of the major identified pathways of early and delayed forms of preconditioning. Several autocrine/paracrine mediators released during the period of preconditioning ischaemia act on G-protein coupled receptors and are known to participate in the infarct-limiting effect of ischaemic preconditioning. These include adenosine released during ischaemia as a result of ATP breakdown, bradykinin released from vascular endothelium and mediators of neural origin (noradrenaline and opioid peptides). Reactive oxygen species, especially superoxide anion generated as a result of mitochondrial uncoupling, may also act as upstream mediators. A complex signal cascade is activated which involves activation of protein kinase C isoenzymes, tyrosine kinases and mitogen-activated protein kinases. The phosphorylation cascade is thought to result in activation of the ATP-sensitive potassium (KATP) channel on the mitochondrial inner membrane. At present it remains unknown how opening of this channel confers protection during ischaemia. The participation of other ‘cytoprotective' proteins has been proposed, including proteins that suppress or modulate apoptosis and proteins associated with cytoskeletal integrity (αB-crystallin and 27 kDa heat shock protein). *The participation of endogenous NO (of endothelial or neural origin) in initiating the classical preconditioning mechanism may be model specific. Early protection against cell death and infarction is not NO-dependent whereas preconditioning against arrhythmias does involve NO generation. For delayed preconditioning, evidence for the involvement of NO (possibly as a signalling intermediate downstream of bradykinin) is more persuasive and consistent. The distinguishing feature of delayed preconditioning is the co-ordinated regulation of a gene transcription programme as a result of upstream kinase signalling. The delayed phase of protection is dependent on de novo synthesis of inducible proteins. Those thought to be particularly important in the acquisition of delayed tolerance to ischaemia include iNOS, cyclo-oxygenase-2 and intracellular antioxidant enzymes such as manganese – superoxide dismutase. For more detailed discussion see Baxter & Ferdinandy (2001).
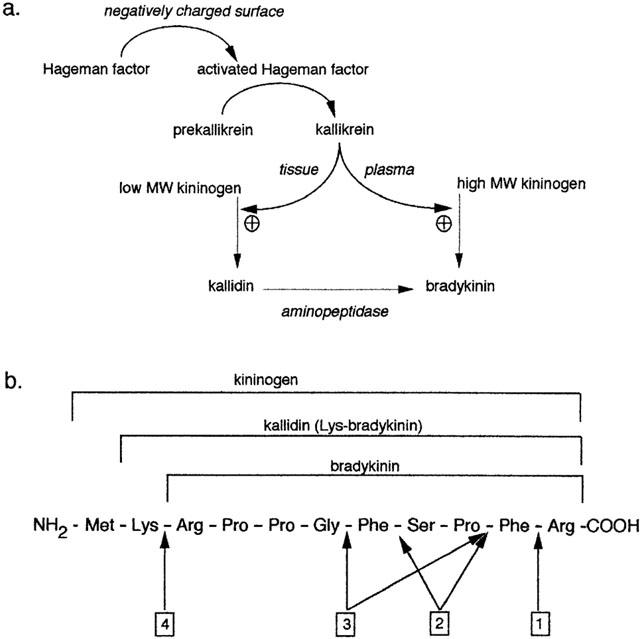
(a) Synthesis of bradykinin. The activated form of Hageman factor promotes the conversion of prekallikrein to kallikrein. In rat, both plasma and tissue kallikrein catalyse the formation of bradykinin. In humans, however, plasma kallikrein generates bradykinin using high molecular weight kininogens and tissue kallikrein generates kallidin using low molecular weight kininogens. (b) Basic amino acid sequence of bradykinin and precursors. Arrows designate the peptide bonds cleaved by: 1 kininase-I; 2 angiotensin converting enzyme (kininase-II); 3 neutral endopeptidase; 4 aminopeptidase.
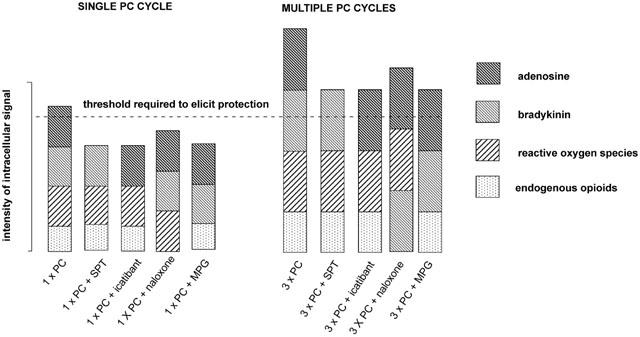
Schematic illustrating the multiple trigger hypothesis of classical preconditioning advanced by Goto et al. (1995). The hypothesis proposes that the preconditioning response is only elicited when a critical threshold of intracellular kinase activity is exceeded. All of the endogenous mediators known to act as triggers of the preconditioning response can independently activate the intracellular signal cascade but they act in concert to trigger the preconditioning response. When a single preconditioning cycle is used (1×PC), blockade of adenosine receptors with 8-sulphophenyltheophylin (SPT), blockade of bradykinin B2 receptors with icatibant, blockade of opioid receptors with (−)-naloxone or scavenging of free radicals with mercaptoprionylglycine (MPG) will be sufficient to reduce the intensity of the intracellular signal below the critical threshold. When multiple preconditioning cycles are used (e.g. 3×PC), relatively more of the endogenous triggers are released. Under these conditions, antagonism of any single trigger may still result in sufficient kinase activation to exceed the threshold which elicits protection.
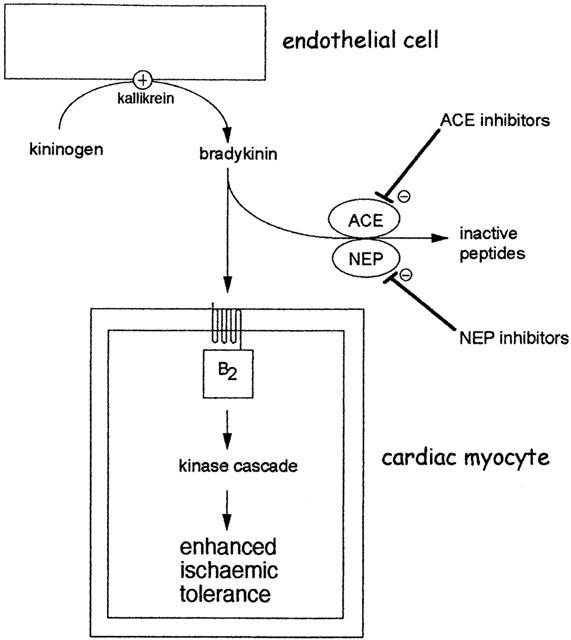
Augmentation of the preconditioning response by angiotensin converting enzyme (ACE) inhibitors or neutral endopeptidase (NEP) inhibitors. Bradykinin is efficiently and rapidly degraded by several enzymes especially ACE and NEP. Inhibition of these enzymes increases the availability of bradykinin at B2 receptors on cardiac myocytes. During very brief periods of ischaemia, interstitial bradykinin concentration may be insufficient to initiate the preconditioning mechanism. However, in the presence of an ACE or NEP inhibitor, augmentation of bradykinin concentration is sufficient to initiate preconditioning. The ability of ACE inhibitors to potentiate subthreshold ischaemic stimuli has been demonstrated for both early and delayed forms of preconditioning (see text).
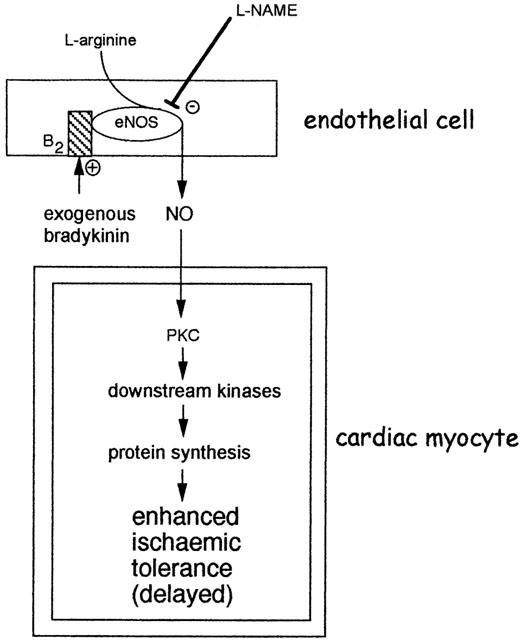
Proposed mechanism for the induction of delayed cardioprotection by bradykinin. The immediate activation of NOS as a result of bradykinin B2 receptor activation leads to the generation of NO. The most likely NOS isoform is eNOS. According to prevailing the delayed preconditioning hypothesis, NO could subsequently trigger an adaptive response in cardiac myocytes, involving the activation of protein kinase C (PKC) isoforms and other kinases. The subsequent induction of unknown mediators of protection results in enhanced tolerance to ischaemia 24 h following liberation or application of bradykinin. Adapted from Ebrahim et al. (2001b).
References
-
- AHMAD M., ZEITLIN I.J., PARRATT J.R. The release of kininase from rat isolated hearts during myocardial ischaemia. Immunopharmacology. 1996;33:299–300. - PubMed
-
- ANDERSON B., KHAPER N., DHALLA A.K., SINGAL P.K. Anti-free radical mechanisms in captopril protection against reperfusion injury in isolated rat hearts. Can. J. Cardiol. 1996;12:1099–1104. - PubMed
-
- ARAMORI I., ZENKOH J., MORIKAWA N., O'DONNELL N., ASANO M., NAKAMURA K., IWAMI M., KOJO H., NOTSU Y. Novel subtype-selective nonpeptide bradykinin receptor antagonists FR167344 and FR173657. Mol. Pharmacol. 1997;51:171–176. - PubMed
-
- ARMSTRONG S., GANOTE C.E. Adenosine receptor specificity in preconditioning of isolated rabbit cardiomyocytes: evidence of A3 receptor involvement. Cardiovasc. Res. 1994;28:1049–1056. - PubMed
-
- ASHER J.R., NAFTILAN A.J. Vasopeptidase inhibition: a new direction in cardiovascular treatment. Curr. Hypertens. Rep. 2000;2:384–391. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
