

A question of size: the eukaryotic proteome and the problems in defining it
- PMID: 11861898
- PMCID: PMC101239
- DOI: 10.1093/nar/30.5.1083
A question of size: the eukaryotic proteome and the problems in defining it
Abstract
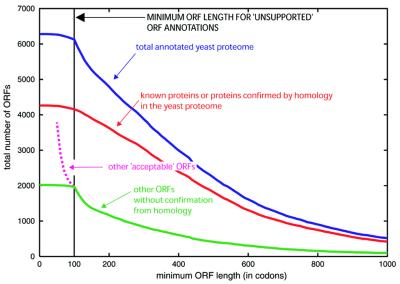
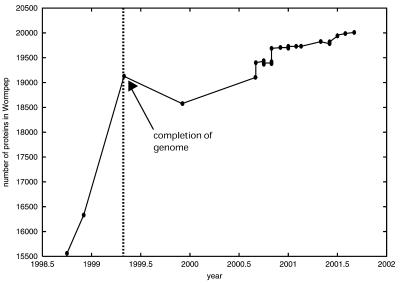
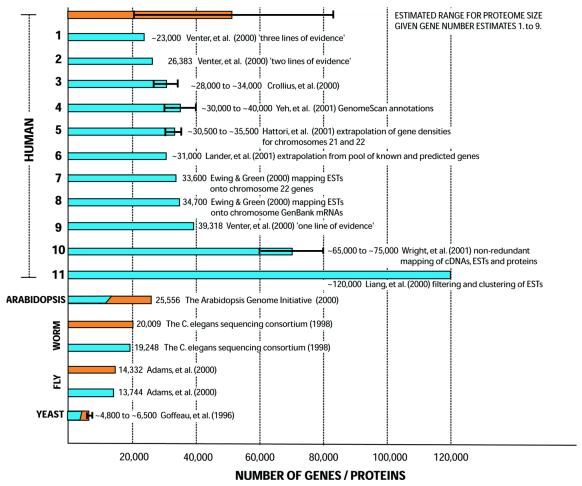
We discuss the problems in defining the extent of the proteomes for completely sequenced eukaryotic organisms (i.e. the total number of protein-coding sequences), focusing on yeast, worm, fly and human. (i) Six years after completion of its genome sequence, the true size of the yeast proteome is still not defined. New small genes are still being discovered, and a large number of existing annotations are being called into question, with these questionable ORFs (qORFs) comprising up to one-fifth of the 'current' proteome. We discuss these in the context of an ideal genome-annotation strategy that considers the proteome as a rigorously defined subset of all possible coding sequences ('the orfome'). (ii) Despite the greater apparent complexity of the fly (more cells, more complex physiology, longer lifespan), the nematode worm appears to have more genes. To explain this, we compare the annotated proteomes of worm and fly, relating to both genome-annotation and genome evolution issues. (iii) The unexpectedly small size of the gene complement estimated for the complete human genome provoked much public debate about the nature of biological complexity. However, in the first instance, for the human genome, the relationship between gene number and proteome size is far from simple. We survey the current estimates for the numbers of human genes and, from this, we estimate a range for the size of the human proteome. The determination of this is substantially hampered by the unknown extent of the cohort of pseudogenes ('dead' genes), in combination with the prevalence of alternative splicing. (Further information relating to yeast is available at http://genecensus.org/yeast/orfome)
Figures
References
-
- Petrov D.A. (2001) Evolution of genome size: new approaches to an old problem. Trends Genet., 17, 23–28. - PubMed
-
- Claverie J.M. (2001) What if there are only 30,000 human genes? Science, 291, 1255–1257. - PubMed
-
- Goffeau A., Barrell,B.G, Bussey,H., Davis,R.W., Dujon,B., Feldmann,H., Galibert,F., Hoheisel,J.D, Jacq,C., Johnston,M. et al. (1996) Life with 6000 genes. Science, 274, 546, 563-567. - PubMed
-
- Dujon B. (1996) The yeast genome project: what did we learn? Trends Genet., 12, 263–270. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
