

BALSA: Bayesian algorithm for local sequence alignment
- PMID: 11861921
- PMCID: PMC101229
- DOI: 10.1093/nar/30.5.1268
BALSA: Bayesian algorithm for local sequence alignment
Abstract
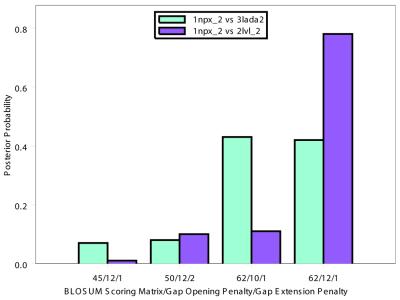
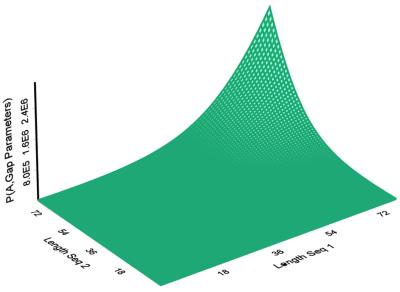
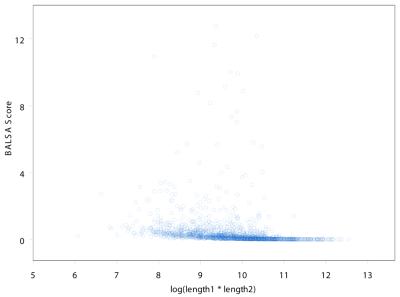
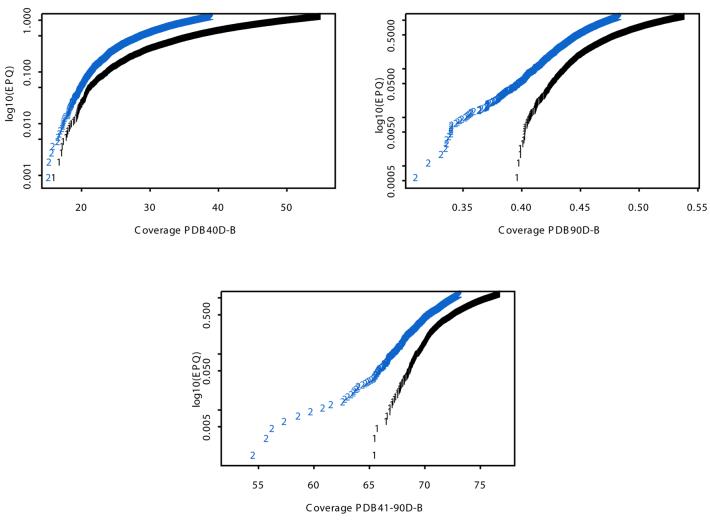
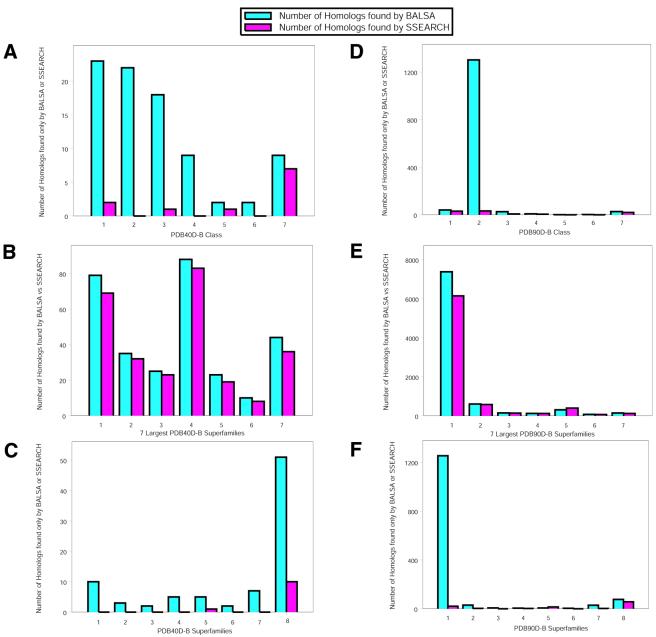
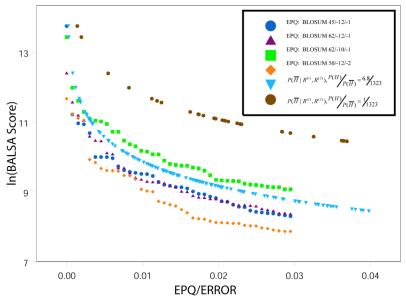
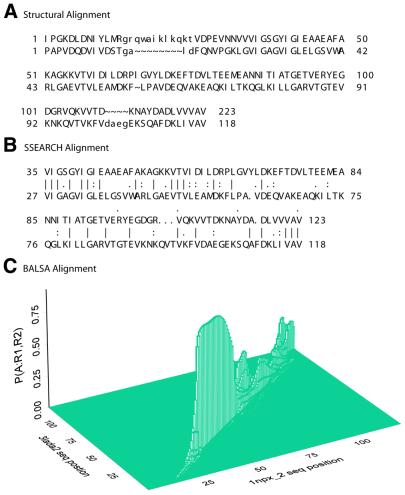
The Smith-Waterman algorithm yields a single alignment, which, albeit optimal, can be strongly affected by the choice of the scoring matrix and the gap penalties. Additionally, the scores obtained are dependent upon the lengths of the aligned sequences, requiring a post-analysis conversion. To overcome some of these shortcomings, we developed a Bayesian algorithm for local sequence alignment (BALSA), that takes into account the uncertainty associated with all unknown variables by incorporating in its forward sums a series of scoring matrices, gap parameters and all possible alignments. The algorithm can return both the joint and the marginal optimal alignments, samples of alignments drawn from the posterior distribution and the posterior probabilities of gap penalties and scoring matrices. Furthermore, it automatically adjusts for variations in sequence lengths. BALSA was compared with SSEARCH, to date the best performing dynamic programming algorithm in the detection of structural neighbors. Using the SCOP databases PDB40D-B and PDB90D-B, BALSA detected 19.8 and 41.3% of remote homologs whereas SSEARCH detected 18.4 and 38% at an error rate of 1% errors per query over the databases, respectively.
Figures
References
-
- Smith T.F. and Waterman,M.S. (1981) Identification of common molecular subsequences. J. Mol. Biol., 147, 195–197. - PubMed
-
- Needleman S.B. and Wunsch,C.D. (1970) a general method applicable to the search for similarities in the amino acid sequence of two proteins. J. Mol. Biol., 48, 443–453. - PubMed
-
- Bucher P. and Hofmann,K. (1996) A sequence similarity search algorithm based on a probabilistic interpretation of an alignment scoring system. Proc. Int. Conf. Intell. Syst. Mol. Biol., 44, 44–51. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
