

Tumour class prediction and discovery by microarray-based DNA methylation analysis
- PMID: 11861926
- PMCID: PMC101257
- DOI: 10.1093/nar/30.5.e21
Tumour class prediction and discovery by microarray-based DNA methylation analysis
Abstract
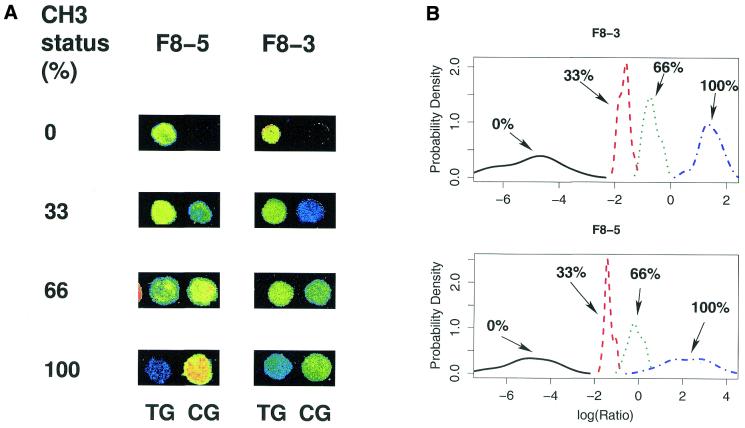
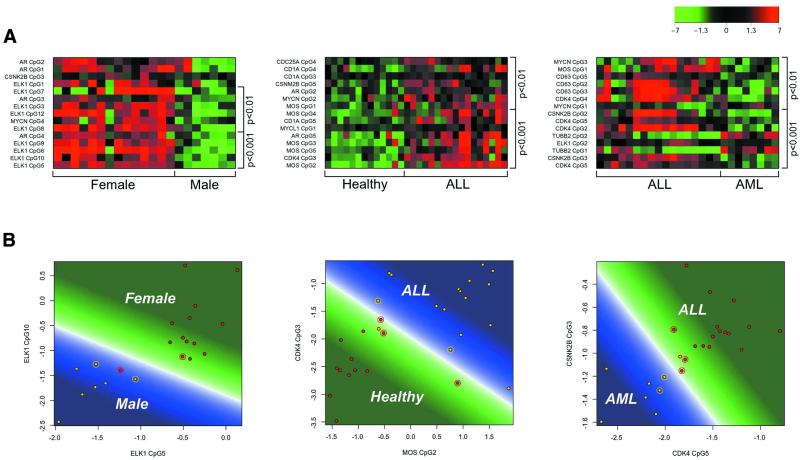
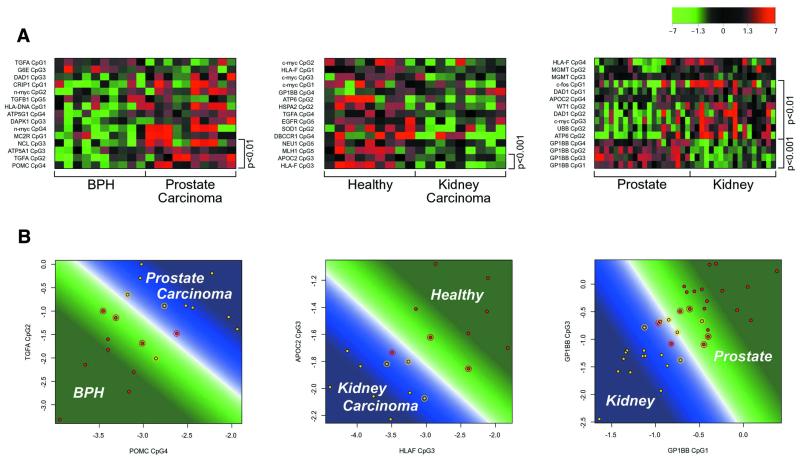
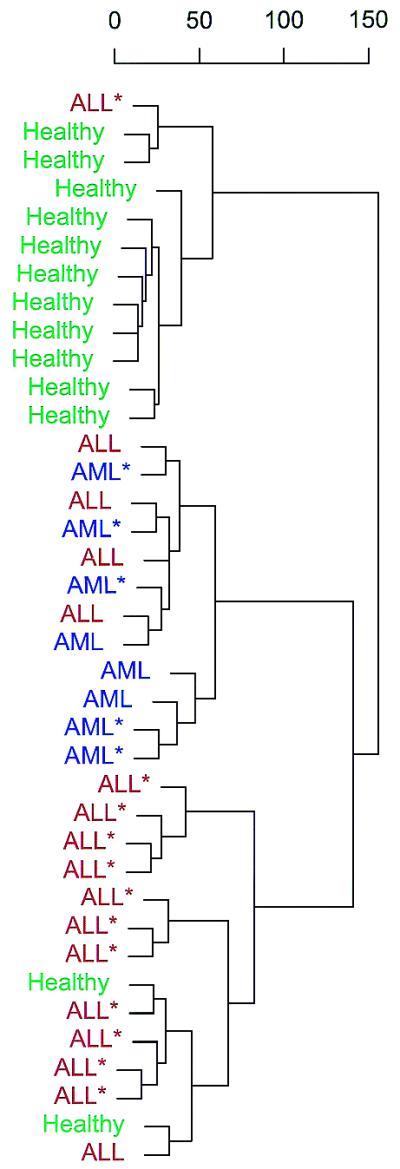
Aberrant DNA methylation of CpG sites is among the earliest and most frequent alterations in cancer. Several studies suggest that aberrant methylation occurs in a tumour type-specific manner. However, large-scale analysis of candidate genes has so far been hampered by the lack of high throughput assays for methylation detection. We have developed the first microarray-based technique which allows genome-wide assessment of selected CpG dinucleotides as well as quantification of methylation at each site. Several hundred CpG sites were screened in 76 samples from four different human tumour types and corresponding healthy controls. Discriminative CpG dinucleotides were identified for different tissue type distinctions and used to predict the tumour class of as yet unknown samples with high accuracy using machine learning techniques. Some CpG dinucleotides correlate with progression to malignancy, whereas others are methylated in a tissue-specific manner independent of malignancy. Our results demonstrate that genome-wide analysis of methylation patterns combined with supervised and unsupervised machine learning techniques constitute a powerful novel tool to classify human cancers.
Figures
References
-
- Jones P.A. (1996) DNA methylation errors and cancer. Cancer Res., 65, 2463–2467. - PubMed
-
- Chan M.F., Liang,G. and Jones,P.A. (2000) Relationship between transcription and DNA methylation. Curr. Top. Microbiol. Immunol., 249, 75–86. - PubMed
-
- Christman J.K., Sheikhnejad,G., Dizik,M., Abileah,S. and Wainfan,E. (1993) Reversibility of changes in nucleic acid methylation and gene expression induced in rat liver by severe dietary methyl deficiency. Carcinogenesis, 14, 551–557. - PubMed
-
- Pogribny I.P., Miller,B.J. and James,S.J. (1997) Alterations in hepatic p53 gene methylation patterns during tumor progression with folate/methyl deficiency in the rat. Cancer Lett., 115, 31–38. - PubMed
-
- Hanada M., Delia,D., Aiello,A., Stadtmauer,E. and Reed,J.C. (1993) bcl-2 gene hypomethylation and high-level expression in B-cell chronic lymphocytic leukemia. Blood, 82, 1820–1828. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
