

Structural views of the ligand-binding cores of a metabotropic glutamate receptor complexed with an antagonist and both glutamate and Gd3+
- PMID: 11867751
- PMCID: PMC122404
- DOI: 10.1073/pnas.052708599
Structural views of the ligand-binding cores of a metabotropic glutamate receptor complexed with an antagonist and both glutamate and Gd3+
Abstract
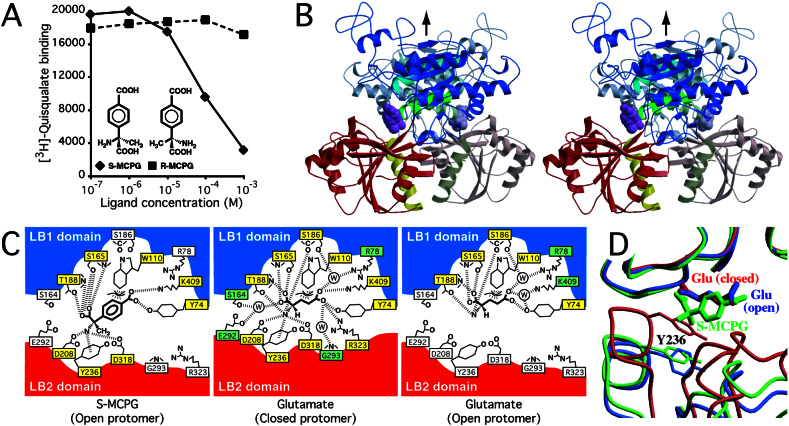
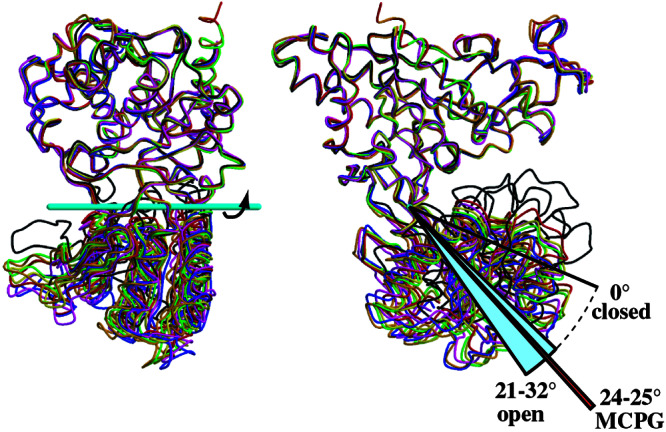
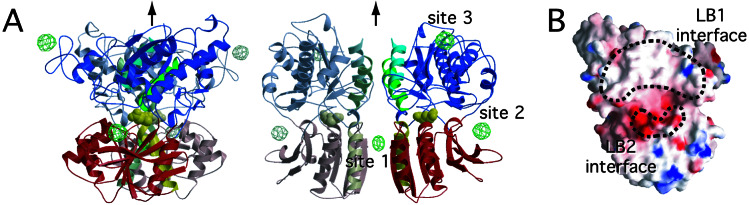
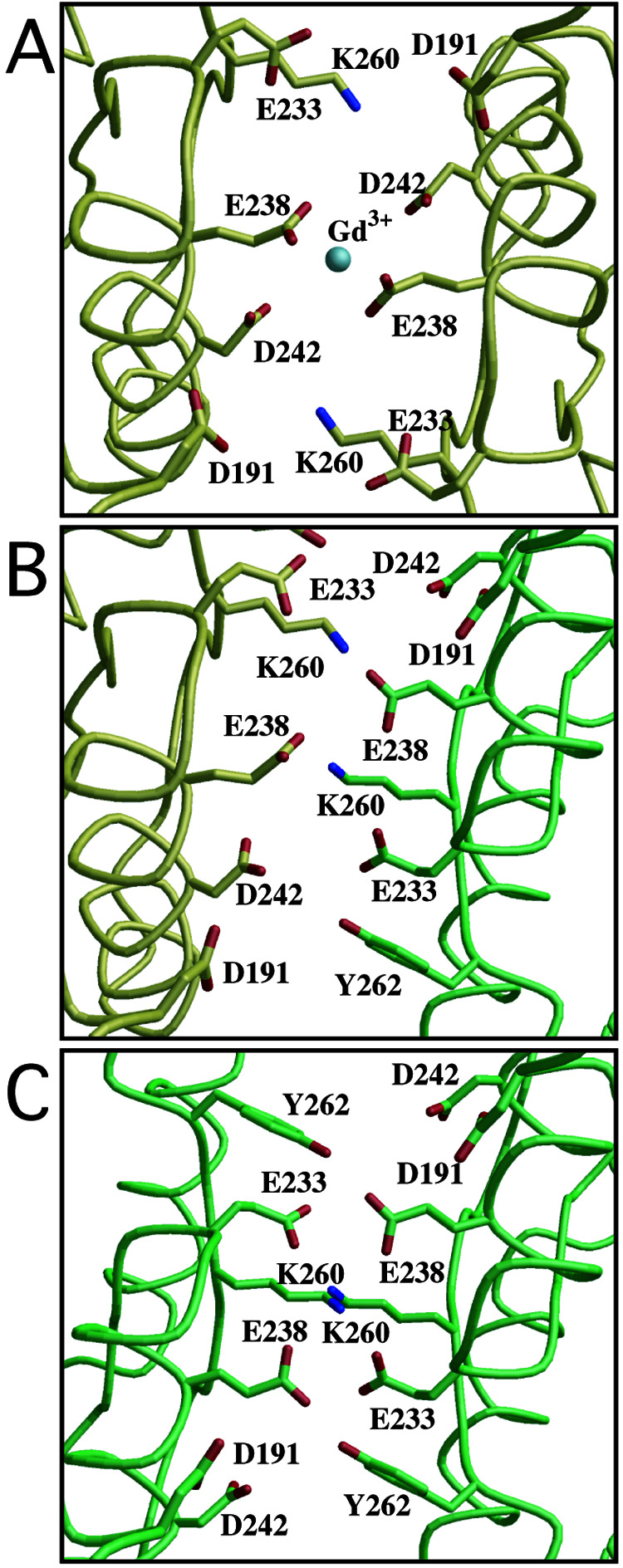
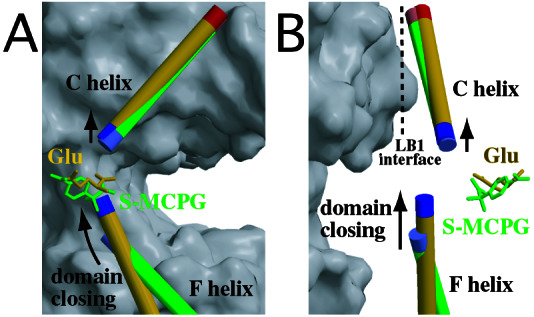
Crystal structures of the extracellular ligand-binding region of the metabotropic glutamate receptor, complexed with an antagonist, (S)-(alpha)-methyl-4-carboxyphenylglycine, and with both glutamate and Gd3+ ion, have been determined by x-ray crystallographic analyses. The structure of the complex with the antagonist is similar to that of the unliganded resting dimer. The antagonist wedges the protomer to maintain an inactive open form. The glutamate/Gd3+ complex is an exact 2-fold symmetric dimer, where each bi-lobed protomer adopts the closed conformation. The surface of the C-terminal domain contains an acidic patch, whose negative charges are alleviated by the metal cation to stabilize the active dimeric structure. The structural comparison between the active and resting dimers suggests that glutamate binding tends to induce domain closing and a small shift of a helix in the dimer interface. Furthermore, an interprotomer contact including the acidic patch inhibited dimer formation by the two open protomers in the active state. These findings provide a structural basis to describe the link between ligand binding and the dimer interface.
Figures
References
-
- Masu M, Tanabe Y, Tsuchida K, Shigemoto R, Nakanishi S. Nature (London) 1991;349:760–765. - PubMed
-
- Nakanishi S, Masu M. Annu Rev Biophys Biomol Struct. 1994;23:319–348. - PubMed
-
- Hollmann M, Heinemann S. Annu Rev Neurosci. 1994;17:31–108. - PubMed
-
- Kunishima N, Shimada Y, Tsuji Y, Sato T, Yamamoto M, Kumasaka T, Nakanishi S, Jingami H, Morikawa K. Nature (London) 2000;407:971–977. - PubMed
-
- Jane D E, Jones P L, Pook P C-K, Salt T E, Sunter D C, Watkins J C. Neurophamacology. 1993;32:725–727. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
