

Gene3D: structural assignment for whole genes and genomes using the CATH domain structure database
- PMID: 11875040
- PMCID: PMC155287
- DOI: 10.1101/gr.213802
Gene3D: structural assignment for whole genes and genomes using the CATH domain structure database
Abstract
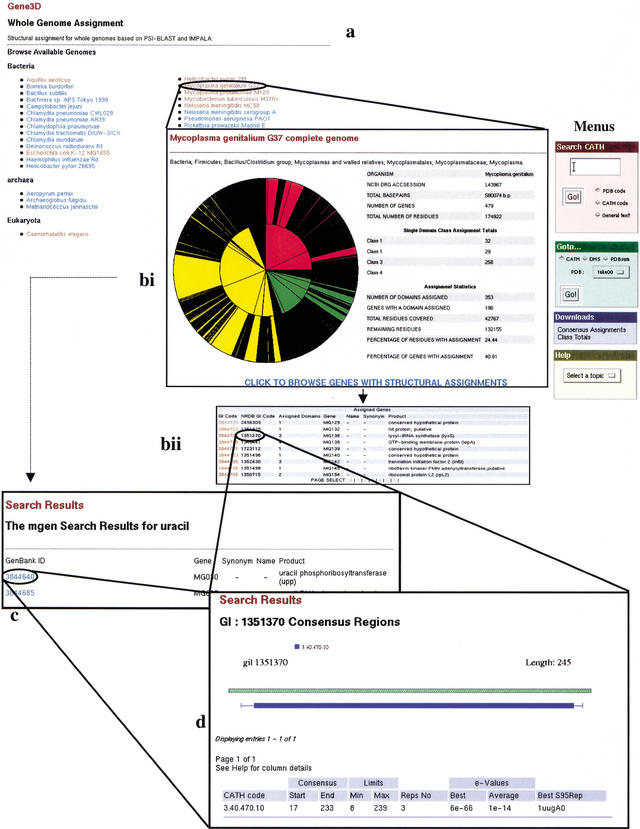
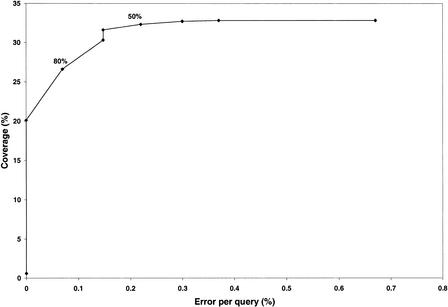
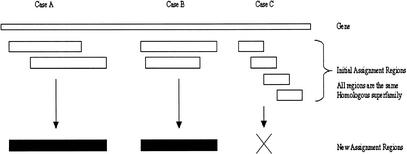
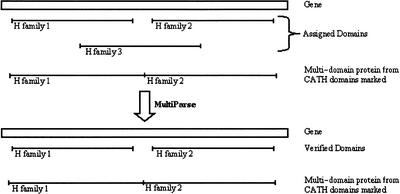
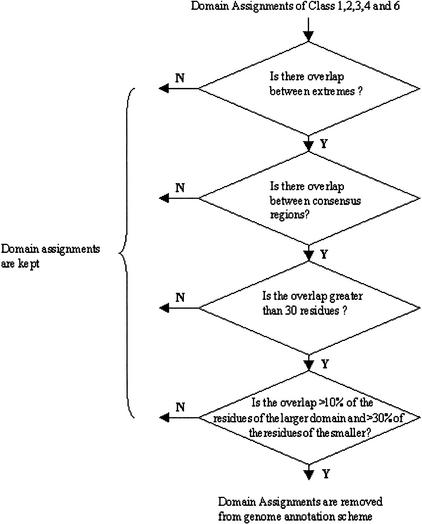
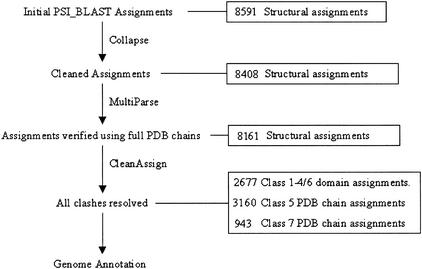
We present a novel web-based resource, Gene3D, of precalculated structural assignments to gene sequences and whole genomes. This resource assigns structural domains from the CATH database to whole genes and links these to their curated functional and structural annotations within the CATH domain structure database, the functional Dictionary of Homologous Superfamilies (DHS) and PDBsum. Currently Gene3D provides annotation for 36 complete genomes (two eukaryotes, six archaea, and 28 bacteria). On average, between 30% and 40% of the genes of a given genome can be structurally annotated. Matches to structural domains are found using the profile-based method (PSI-BLAST). and a novel protocol, DRange, is used to resolve conflicts in matches involving different homologous superfamilies.
Figures

References
-
- Apweiler R, Biswas M, Fleischmann W, Kanapin A, Karavidopoulou Y, Kersey P, Kriventseva EV, Mittard V, Mulder N, Phan, et al. Proteome Analysis Database: Online application of InterPro and CluSTr for the functional classification of proteins in whole genomes. Nucleic Acids Res. 2001b;29:44–48. - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Research Materials