

Mutations in the inosine monophosphate dehydrogenase 1 gene (IMPDH1) cause the RP10 form of autosomal dominant retinitis pigmentosa
- PMID: 11875050
- PMCID: PMC2585828
- DOI: 10.1093/hmg/11.5.559
Mutations in the inosine monophosphate dehydrogenase 1 gene (IMPDH1) cause the RP10 form of autosomal dominant retinitis pigmentosa
Abstract
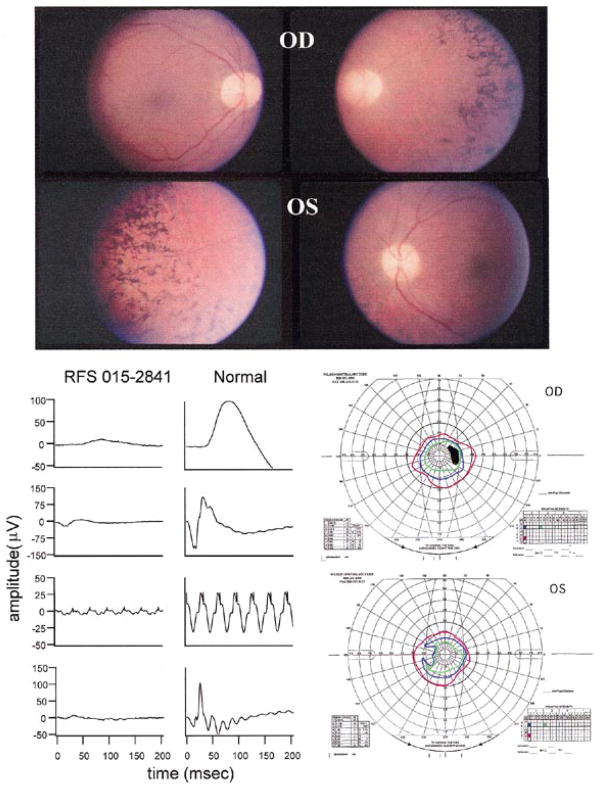
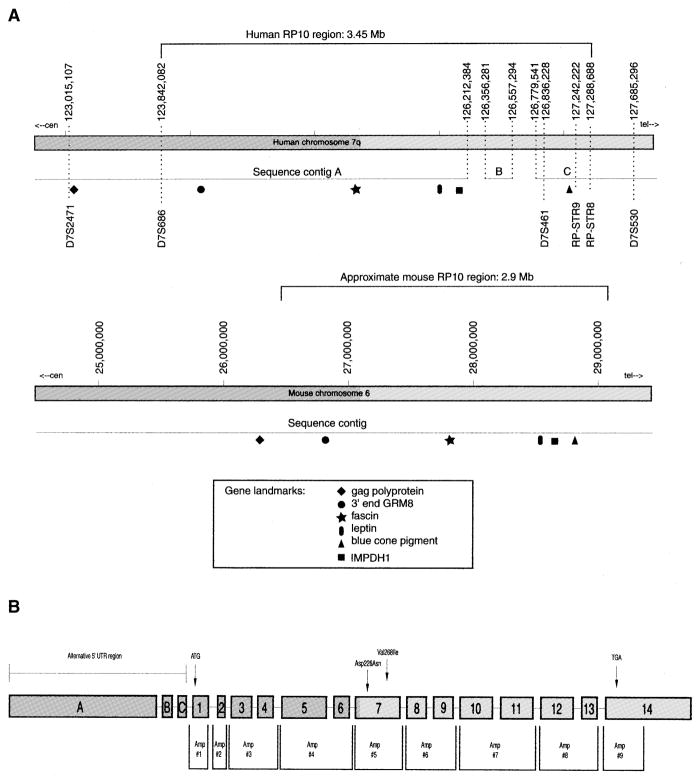
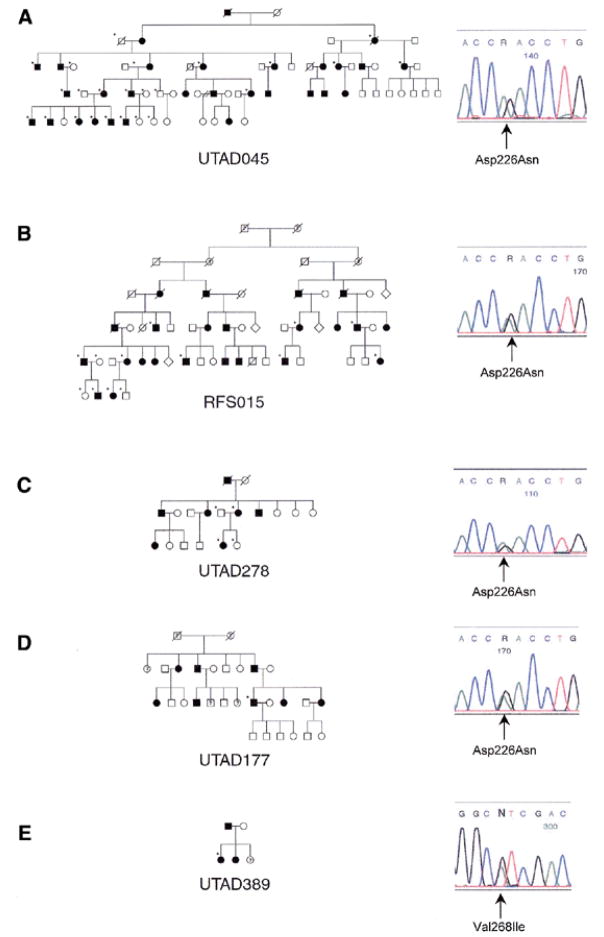
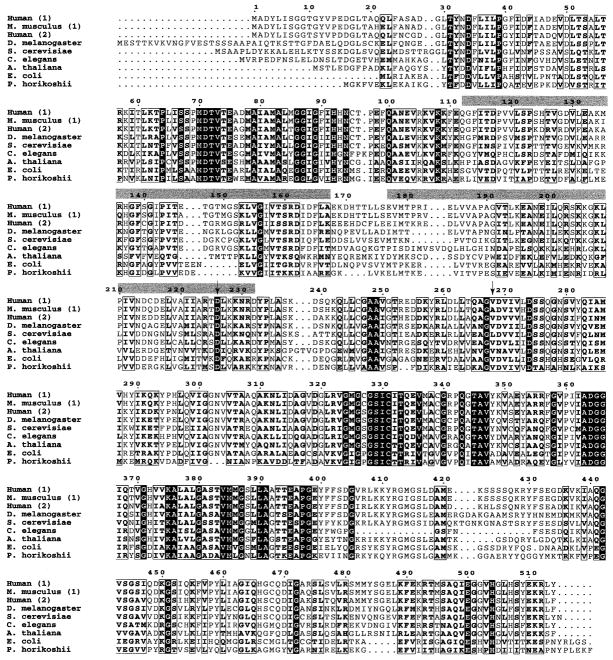
Autosomal dominant retinitis pigmentosa (adRP) is a heterogeneous set of progressive retinopathies caused by several distinct genes. One locus, the RP10 form of adRP, maps to human chromosome 7q31.1 and may account for 5-10% of adRP cases among Americans and Europeans. We identified two American families with the RP10 form of adRP by linkage mapping and used these families to reduce the linkage interval to 3.45 Mb between the flanking markers D7S686 and RP-STR8. Sequence and transcript analysis identified 54 independent genes within this region, at least 10 of which are retinal-expressed and thus candidates for the RP10 gene. A screen of retinal transcripts comparing retinas from normal mice to retinas from crx-/crx- knockout mice (with poorly differentiated photoreceptors) demonstrated a 6-fold reduction in one candidate, inosine monophosphate dehydrogenase 1 (IMPDH1; EC 1.1.1.205). Since many of the genes known to cause retinitis pigmentosa are under CRX control in photoreceptors, IMPDH1 became a high-priority candidate for mutation screening. DNA sequencing of affected individuals from the two American RP10 families revealed a GAC-->AAC transition in codon 226 substituting an asparagine for an aspartic acid in both families. The identical mutation was also found in a British RP10 family. The Asp226Asn missense mutation is present in all affected individuals tested and absent from unaffected controls. The aspartic acid at codon 226 is conserved in all IMPDH genes, in all species examined, including bacteria, suggesting that this mutation is highly deleterious. Subsequent screening of probands from 60 other adRP families revealed an additional family with this mutation, confirming its association with retinitis pigmentosa and the relatively high frequency of this mutation. Another IMPDH1 substitution, Val268Ile, was also observed in this cohort of patients but not in controls. IMPDH1 is a ubiquitously expressed enzyme, functioning as a homotetramer, which catalyzed the rate-limiting step in de novo synthesis of guanine nucleotides. As such, it plays an important role in cyclic nucleoside metabolism within photoreceptors. Several classes of drugs are known to affect IMPDH isoenzymes, including nucleotide and NAD analogs, suggesting that small-molecule therapy may be available, one day, for RP10 patients.
Figures
References
-
- Heckenlively JR, Daiger SP. Hereditary retinal and choroidal degenerations. In: Rimoin DI, Connor JM, Pyeritz RE, editors. Principals and Practices of Medical Genetics. 4. Vol. 1. Churchill Livingston; New York: 2001. pp. 2255–2576.
-
- Jordan SA, Farrar GJ, Kenna P, Humphries MM, Sheils DM, Kumar-Singh R, Sharp EM, Soriano N, Ayuso C, Benitez J, et al. Localization of an autosomal dominant retinitis pigmentosa gene to chromosome 7q. Nat Genet. 1993;4:54–58. - PubMed
-
- McGuire RE, Gannon AM, Sadler-Sullivan LA, Rodriguez JA, Daiger SP. Evidence for a major gene (RP10) for autosomal dominant retinitis pigmentosa on chromosome 7q: linkage mapping in a second, unrelated family. Hum Genet. 1995;95:71–74. - PubMed
-
- McGuire RE, Jordan SA, Braden VV, Bouffard GG, Humphries P, Green ED, Daiger SP. Mapping the RP10 locus for autosomal dominant retinitis pigmentosa on 7q: refined genetic positioning and localization within a well-defined YAC contig. Genome Res. 1996;6:255–266. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
