

Re-annotation of genome microbial coding-sequences: finding new genes and inaccurately annotated genes
- PMID: 11879526
- PMCID: PMC77393
- DOI: 10.1186/1471-2105-3-5
Re-annotation of genome microbial coding-sequences: finding new genes and inaccurately annotated genes
Abstract
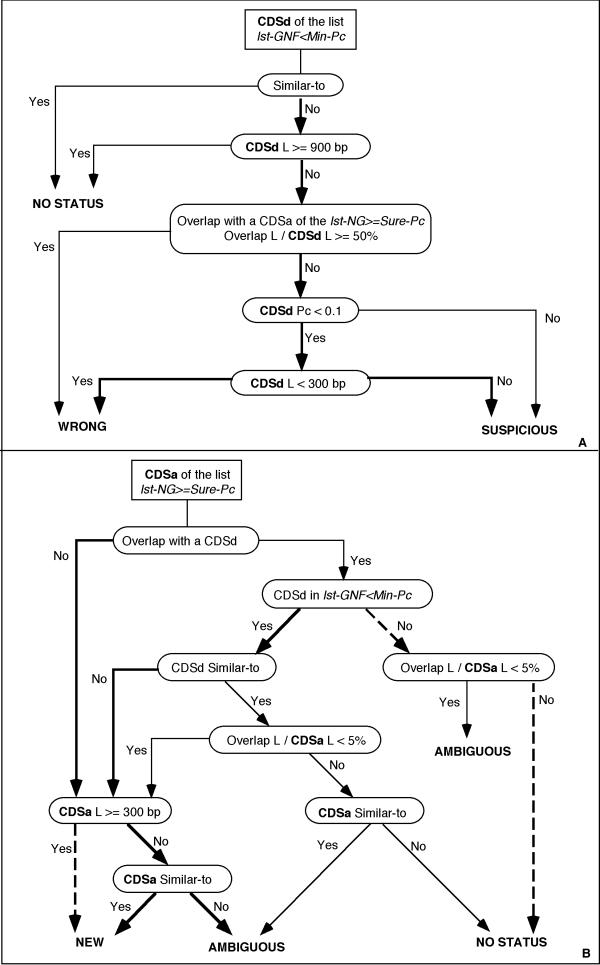
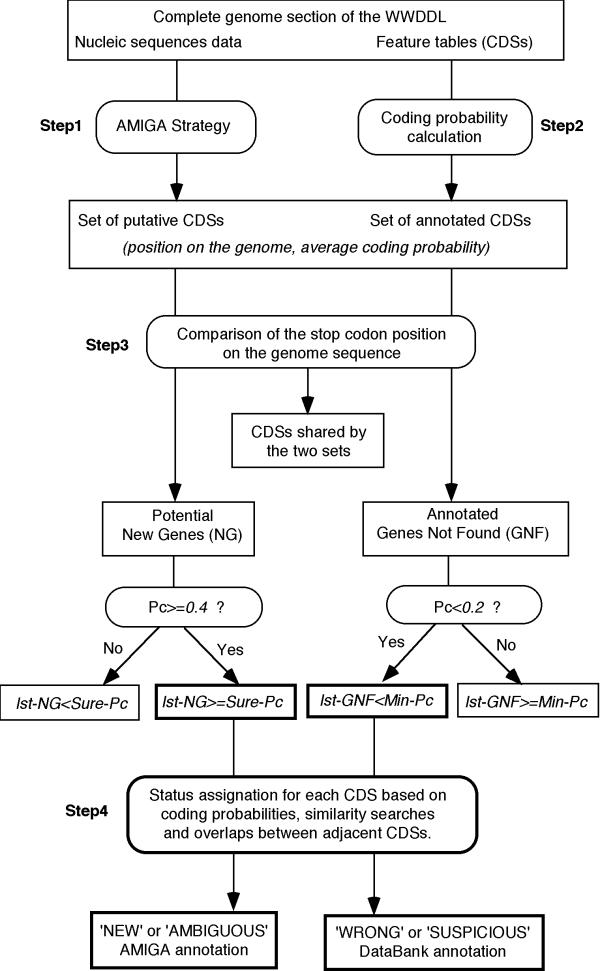
Background: Analysis of any newly sequenced bacterial genome starts with the identification of protein-coding genes. Despite the accumulation of multiple complete genome sequences, which provide useful comparisons with close relatives among other organisms during the annotation process, accurate gene prediction remains quite difficult. A major reason for this situation is that genes are tightly packed in prokaryotes, resulting in frequent overlap. Thus, detection of translation initiation sites and/or selection of the correct coding regions remain difficult unless appropriate biological knowledge (about the structure of a gene) is imbedded in the approach.
Results: We have developed a new program that automatically identifies biologically significant candidate genes in a bacterial genome. Twenty-six complete prokaryotic genomes were analyzed using this tool, and the accuracy of gene finding was assessed by comparison with existing annotations. This analysis revealed that, despite the enormous effort of genome program annotators, a small but not negligible number of genes annotated within the framework of sequencing projects are likely to be partially inaccurate or plainly wrong. Moreover, the analysis of several putative new genes shows that, as expected, many short genes have escaped annotation. In most cases, these new genes revealed frameshifts that could be either artifacts or genuine frameshifts. Some entirely unexpected new genes have also been identified. This allowed us to get a more complete picture of prokaryotic genomes. The results of this procedure are progressively integrated into the SWISS-PROT reference databank.
Conclusions: The results described in the present study show that our procedure is very satisfactory in terms of gene finding accuracy. Except in few cases, discrepancies between our results and annotations provided by individual authors can be accounted for by the nature of each annotation process or by specific characteristics of some genomes. This stresses that close cooperation between scientists, regular update and curation of the findings in databases are clearly required to reduce the level of errors in genome annotation (and hence in reducing the unfortunate spreading of errors through centralized data libraries).
Figures
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
