

Real-time PCR in virology
- PMID: 11884626
- PMCID: PMC101343
- DOI: 10.1093/nar/30.6.1292
Real-time PCR in virology
Abstract
The use of the polymerase chain reaction (PCR) in molecular diagnostics has increased to the point where it is now accepted as the gold standard for detecting nucleic acids from a number of origins and it has become an essential tool in the research laboratory. Real-time PCR has engendered wider acceptance of the PCR due to its improved rapidity, sensitivity, reproducibility and the reduced risk of carry-over contamination. There are currently five main chemistries used for the detection of PCR product during real-time PCR. These are the DNA binding fluorophores, the 5' endonuclease, adjacent linear and hairpin oligoprobes and the self-fluorescing amplicons, which are described in detail. We also discuss factors that have restricted the development of multiplex real-time PCR as well as the role of real-time PCR in quantitating nucleic acids. Both amplification hardware and the fluorogenic detection chemistries have evolved rapidly as the understanding of real-time PCR has developed and this review aims to update the scientist on the current state of the art. We describe the background, advantages and limitations of real-time PCR and we review the literature as it applies to virus detection in the routine and research laboratory in order to focus on one of the many areas in which the application of real-time PCR has provided significant methodological benefits and improved patient outcomes. However, the technology discussed has been applied to other areas of microbiology as well as studies of gene expression and genetic disease.
Figures
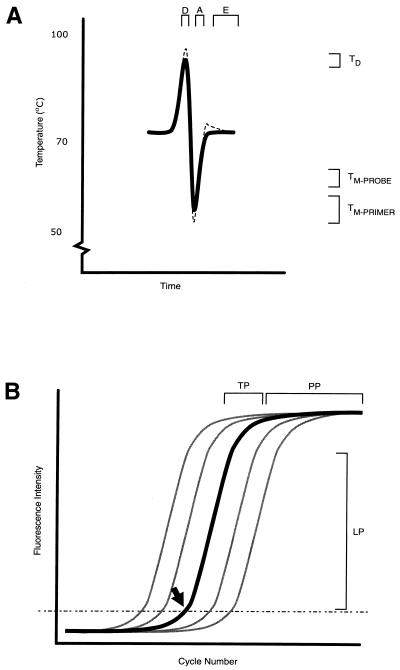
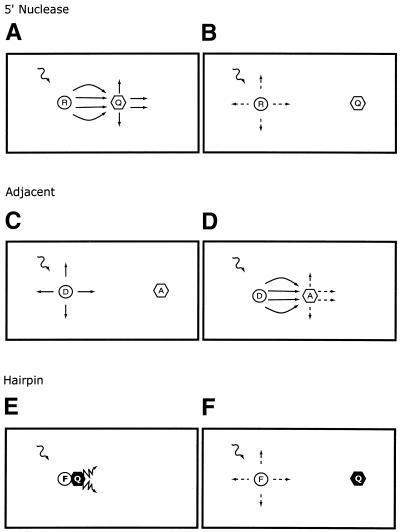
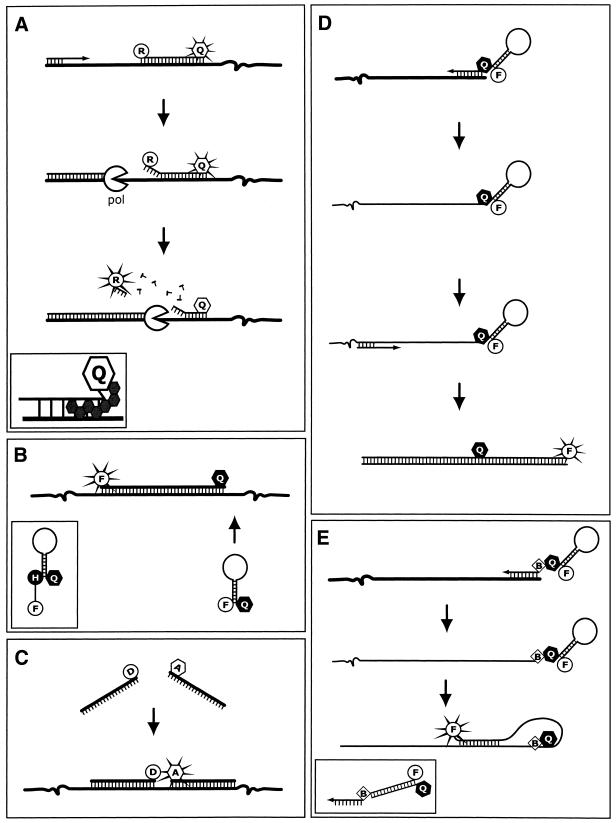
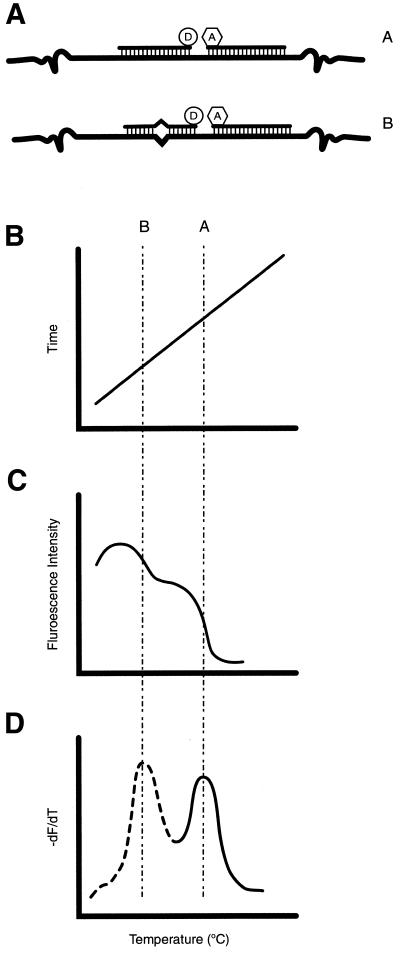
References
-
- Mullis K.B. and Faloona,F. (1987) Specific synthesis of DNA in vitro via a polymerase-catalysed chain reaction. Methods Enzymol., 155, 335–350. - PubMed
-
- Kidd I.M., Clark,D.A. and Emery,V.C. (2000) A non-radioisotopic quantitative competitive polymerase chain reaction method: application in measurement of human herpesvirus 7 load. J. Virol. Methods, 87, 177–181. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
