

The nucleotide sequence of Shiga toxin (Stx) 2e-encoding phage phiP27 is not related to other Stx phage genomes, but the modular genetic structure is conserved
- PMID: 11895953
- PMCID: PMC127862
- DOI: 10.1128/IAI.70.4.1896-1908.2002
The nucleotide sequence of Shiga toxin (Stx) 2e-encoding phage phiP27 is not related to other Stx phage genomes, but the modular genetic structure is conserved
Erratum in
- Infect Immun 2002 Aug;70(8):4755
Abstract
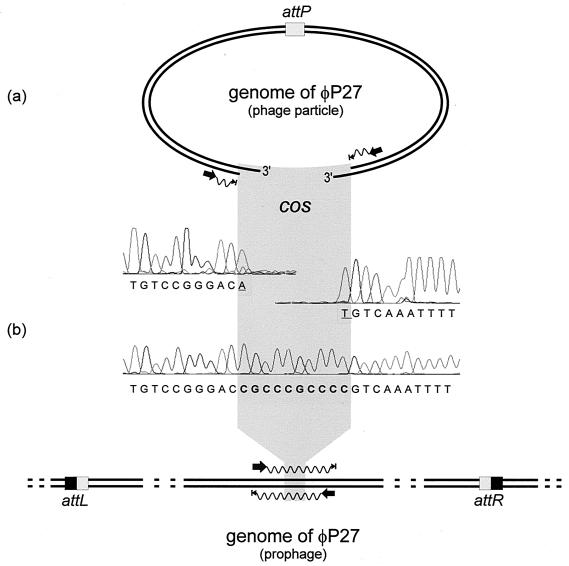
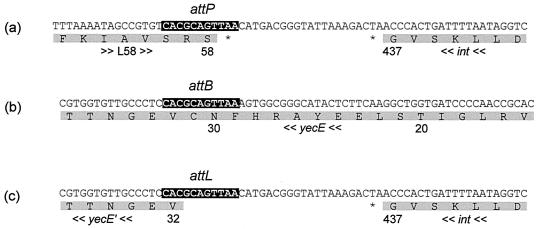
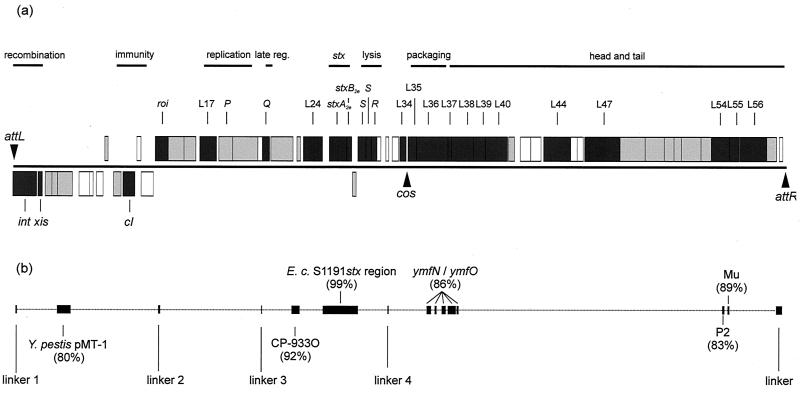
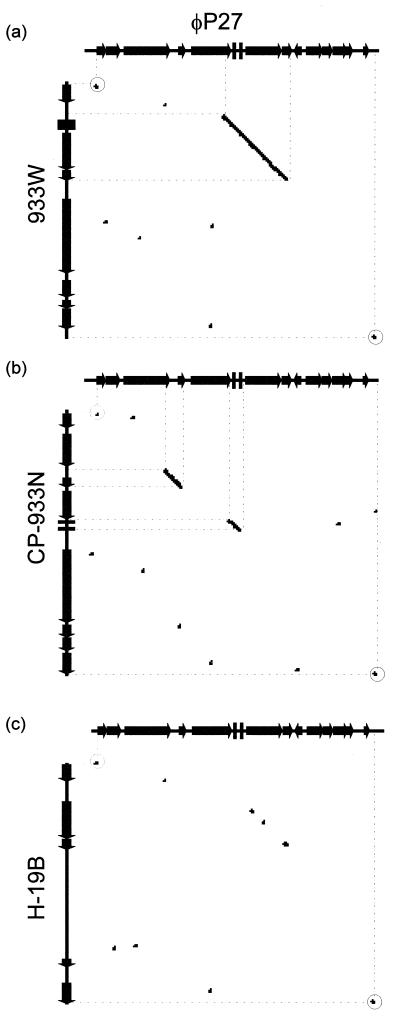
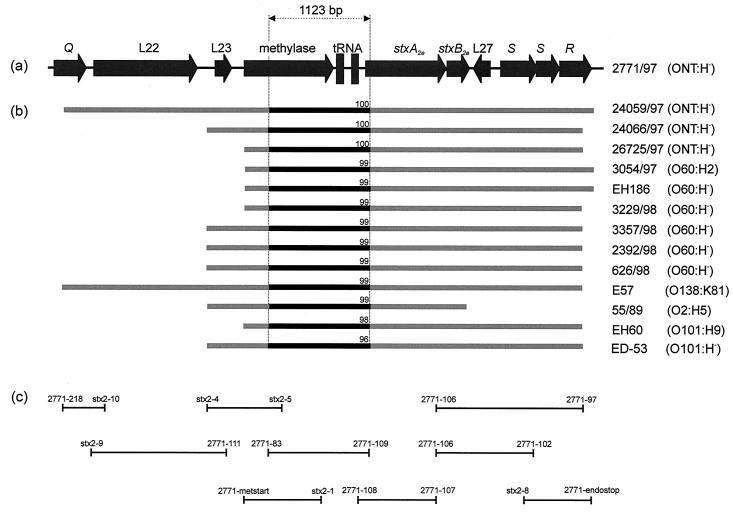
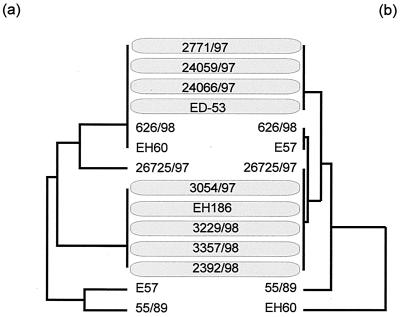
In this study we determined the complete nucleotide sequence of Shiga toxin 2e-encoding bacteriophage phi P27, isolated from the Shiga toxin-producing Escherichia coli patient isolate 2771/97. phi P27 is integrated as a prophage in the chromosomal yecE gene. This integration generates identity segments of attL and attR sites with lengths of 11 nucleotides. The integrated prophage genome has a size of 42,575 bp. We identified 58 open reading frames (ORFs), each with a length of >150 nucleotides. The deduced proteins of 44 ORFs showed significant homologies to other proteins present in sequence databases, whereas 14 putative proteins did not. For 29 proteins, we could deduce a putative function. Most of these are related to the basic phage propagation cycle. The phi P27 genome represents a mosaic composed of genetic elements which are obviously derived from related and unrelated phages. We identified five short linker sequences of 22 to 151 bp in the phi P27 sequence which have also been detected in a couple of other lambdoid phages. These linkers are located between functional modules in the phage genome and are thought to play a role in genetic recombination. Although the overall DNA sequence of phi P27 is not highly related to other known phages, the data obtained demonstrate a typical lambdoid genome structure.
Figures
References
-
- Altschul, S. F., W. Gish, W. Miller, E. W. Myers, and D. J. Lipman. 1990. Basic local alignment search tool. J. Mol. Biol. 215:403-410. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
