

DNA-dependent protein kinase suppresses double-strand break-induced and spontaneous homologous recombination
- PMID: 11904432
- PMCID: PMC122597
- DOI: 10.1073/pnas.052545899
DNA-dependent protein kinase suppresses double-strand break-induced and spontaneous homologous recombination
Abstract
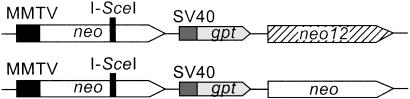
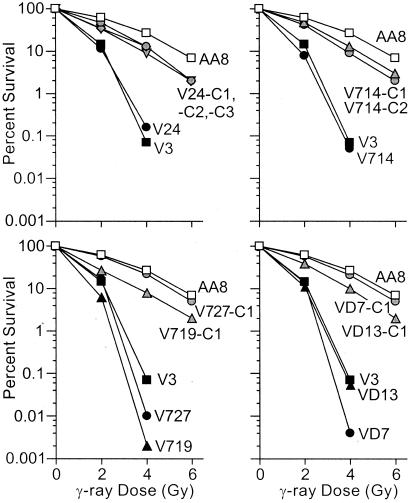
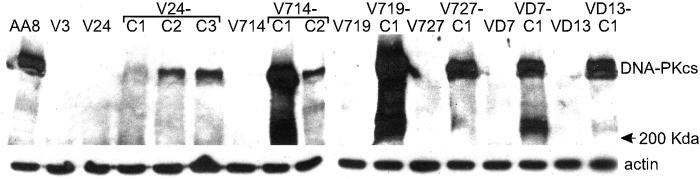
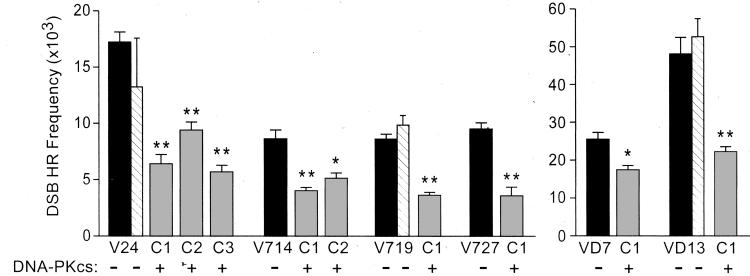
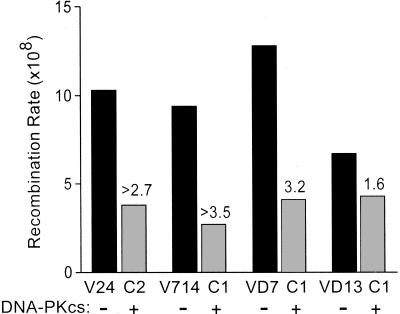
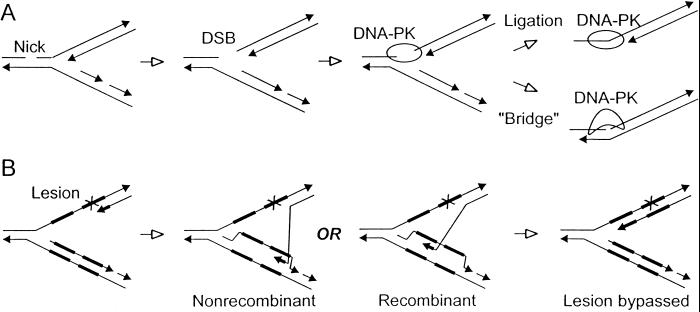
DNA-dependent protein kinase (DNA-PK), composed of Ku70, Ku80, and the catalytic subunit (DNA-PKcs), is involved in repairing double-strand breaks (DSBs) by nonhomologous end-joining (NHEJ). Certain proteins involved in NHEJ are also involved in DSB repair by homologous recombination (HR). To test the effects of DNA-PKcs on DSB-induced HR, we integrated neo direct repeat HR substrates carrying the I-SceI recognition sequence into DNA-PKcs-defective Chinese hamster ovary (V3) cells. The DNA-PKcs defect was complemented with a human DNA-PKcs cDNA. DSB-induced HR frequencies were 1.5- to 3-fold lower with DNA-PKcs complementation. In complemented and uncomplemented strains, all products arose by gene conversion without associated crossover, and average conversion tract lengths were similar. Suppression of DSB-induced HR in complemented cells probably reflects restoration of NHEJ, consistent with competition between HR and NHEJ during DSB repair. Interestingly, spontaneous HR rates were 1.6- to >3.5-fold lower with DNA-PKcs complementation. DNA-PKcs may suppress spontaneous HR through NHEJ of spontaneous DSBs, perhaps at stalled or blocked replication forks. Because replication protein A (RPA) is involved in both replication and HR, and is phosphorylated by DNA-PKcs, it is possible that the suppression of spontaneous HR by DNA-PKcs reflects regulation of replication-dependent HR by DNA-PKcs, perhaps by means of phosphorylation of RPA.
Figures
References
-
- Haber J E. Trends Biochem Sci. 2000;16:259–264.
-
- Nickoloff J A, Singer J D, Hoekstra M F, Heffron F. J Mol Biol. 1989;207:527–541. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
