

Impaired cardiac contractility response to hemodynamic stress in S100A1-deficient mice
- PMID: 11909974
- PMCID: PMC133731
- DOI: 10.1128/MCB.22.8.2821-2829.2002
Impaired cardiac contractility response to hemodynamic stress in S100A1-deficient mice
Abstract
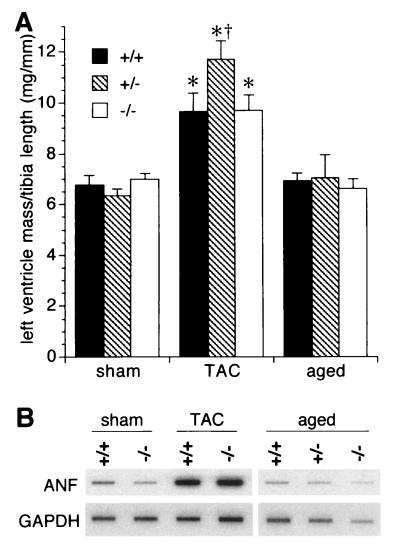
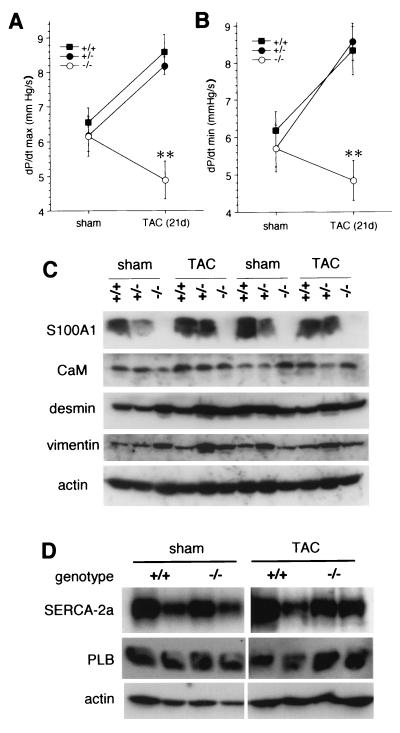
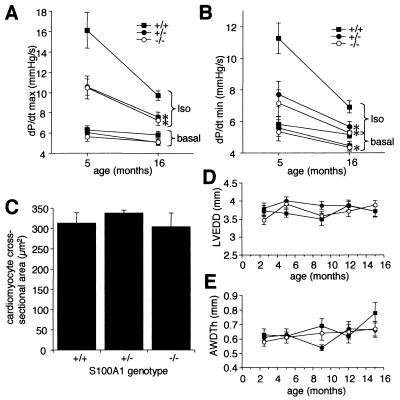
Ca(2+) signaling plays a central role in cardiac contractility and adaptation to increased hemodynamic demand. We have generated mice with a targeted deletion of the S100A1 gene coding for the major cardiac isoform of the large multigenic S100 family of EF hand Ca(2+)-binding proteins. S100A1(-/-) mice have normal cardiac function under baseline conditions but have significantly reduced contraction rate and relaxation rate responses to beta-adrenergic stimulation that are associated with a reduced Ca(2+) sensitivity. In S100A1(-/-) mice, basal left-ventricular contractility deteriorated following 3-week pressure overload by thoracic aorta constriction despite a normal adaptive hypertrophy. Surprisingly, heterozygotes also had an impaired response to acute beta-adrenergic stimulation but maintained normal contractility in response to chronic pressure overload that coincided with S100A1 upregulation to wild-type levels. In contrast to other genetic models with impaired cardiac contractility, loss of S100A1 did not lead to cardiac hypertrophy or dilation in aged mice. The data demonstrate that high S100A1 protein levels are essential for the cardiac reserve and adaptation to acute and chronic hemodynamic stress in vivo.
Figures
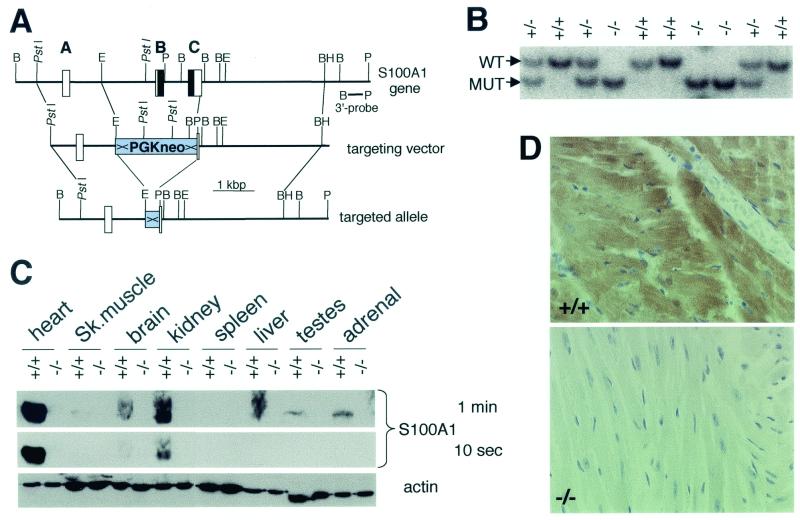
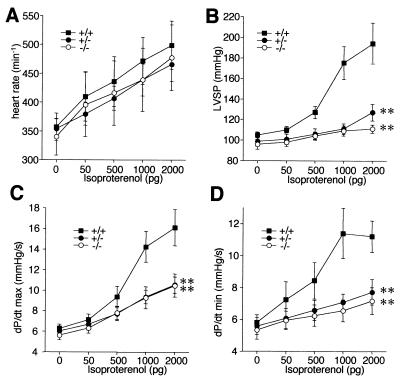
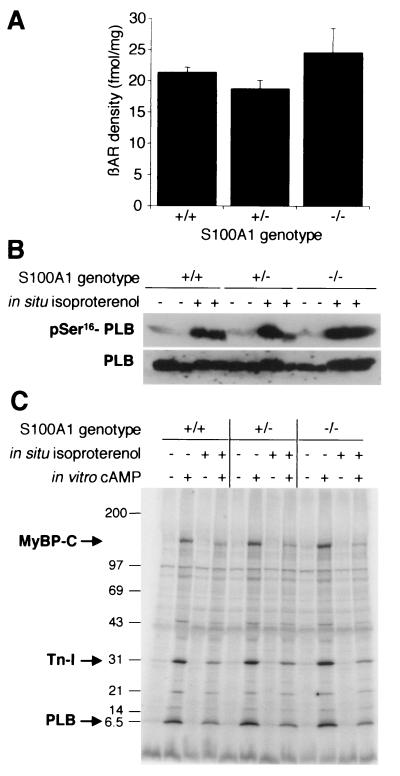
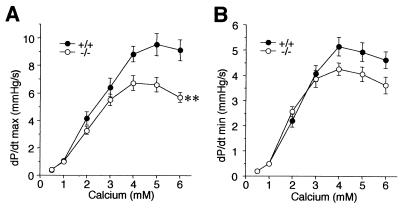
References
-
- Arber, S., J. J. Hunter, J. Ross, M. Hongo, G. Sansig, J. Borg, J. C. Perriard, K. R. Chien, and P. Caroni. 1997. MLP-deficient mice exhibit a disruption of cardiac cytoarchitectural organization, dilated cardiomyopathy, and heart failure. Cell 88:393-403. - PubMed
-
- Baudier, J., E. Bergeret, N. Bertacchi, H. Weintraub, J. Gagnon, and J. Garin. 1995. Interactions of myogenic bHLH transcription factors with calcium-binding calmodulin and S100a (aa) proteins. Biochemistry 34:7834-7846. - PubMed
-
- Chawengsaksophak, K., R. James, V. E. Hammond, F. Köntgen, and F. Beck. 1997. Homeosis and intestinal tumours in Cdx2 mutant mice. Nature 386:84-87. - PubMed
-
- Chien, K. R. 2000. Genomic circuits and the integrative biology of cardiac diseases. Nature 407:227-232. - PubMed
-
- Clapham, D. E. 1995. Calcium signalling. Cell 80:259-268. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous