

Phylogenetic diversity of marine cyanophage isolates and natural virus communities as revealed by sequences of viral capsid assembly protein gene g20
- PMID: 11916671
- PMCID: PMC123904
- DOI: 10.1128/AEM.68.4.1576-1584.2002
Phylogenetic diversity of marine cyanophage isolates and natural virus communities as revealed by sequences of viral capsid assembly protein gene g20
Abstract
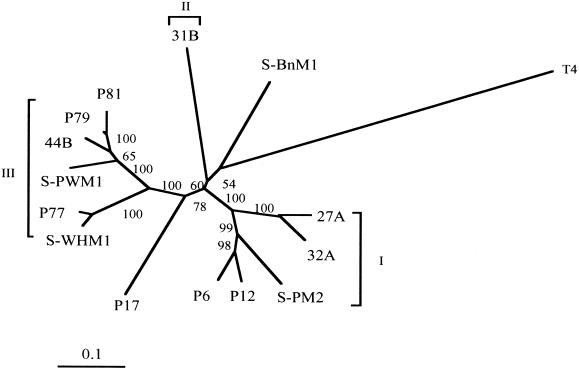
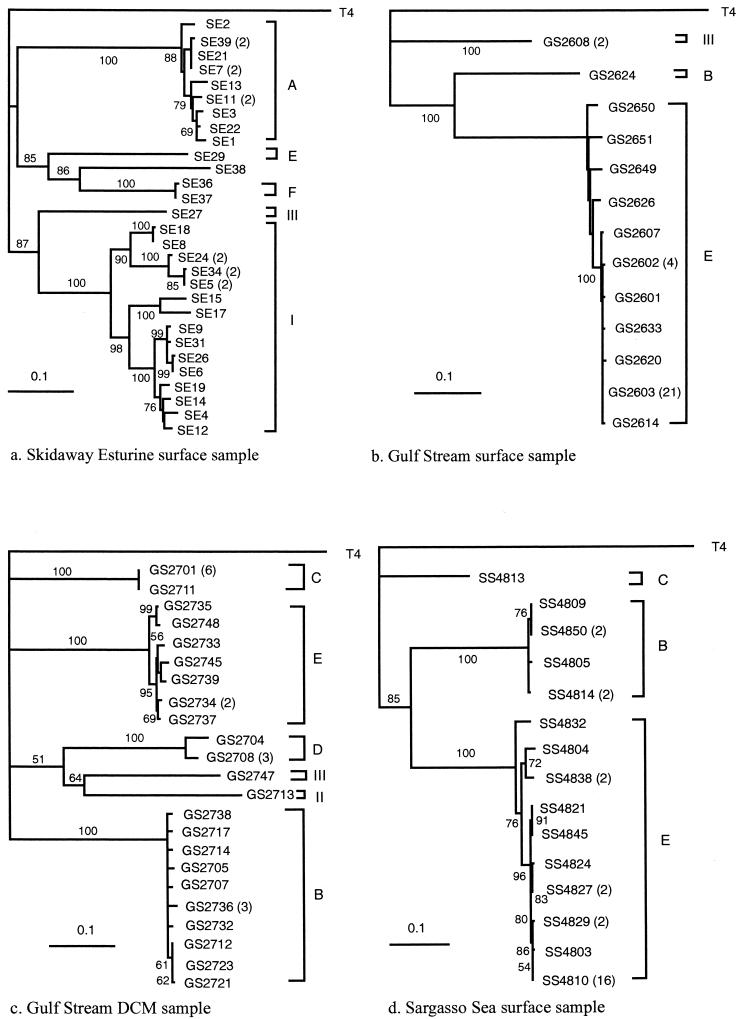
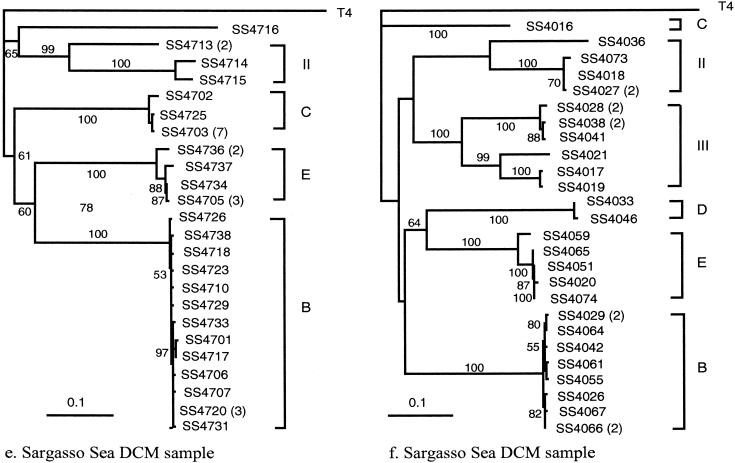
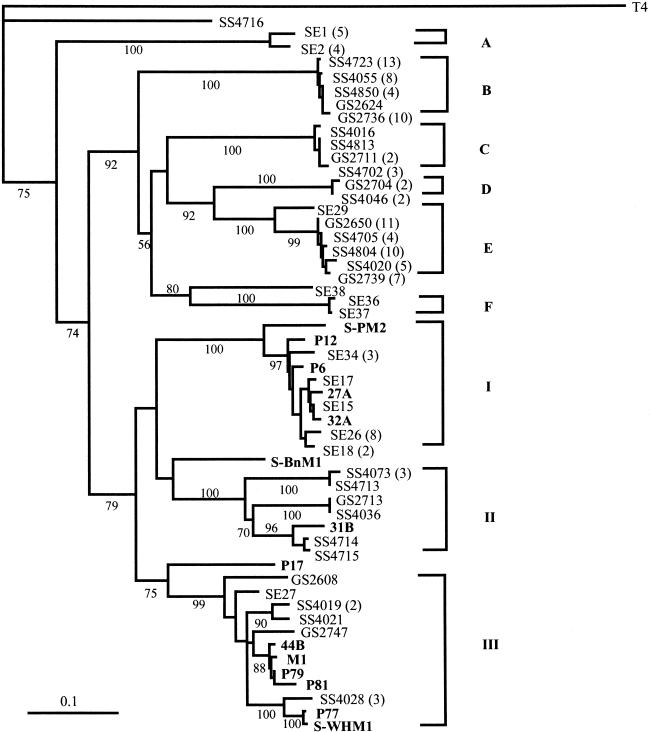
In order to characterize the genetic diversity and phylogenetic affiliations of marine cyanophage isolates and natural cyanophage assemblages, oligonucleotide primers CPS1 and CPS8 were designed to specifically amplify ca. 592-bp fragments of the gene for viral capsid assembly protein g20. Phylogenetic analysis of isolated cyanophages revealed that the marine cyanophages were highly diverse yet more closely related to each other than to enteric coliphage T4. Genetically related marine cyanophage isolates were widely distributed without significant geographic segregation (i.e., no correlation between genetic variation and geographic distance). Cloning and sequencing analysis of six natural virus concentrates from estuarine and oligotrophic offshore environments revealed nine phylogenetic groups in a total of 114 different g20 homologs, with up to six clusters and 29 genotypes encountered in a single sample. The composition and structure of natural cyanophage communities in the estuary and open-ocean samples were different from each other, with unique phylogenetic clusters found for each environment. Changes in clonal diversity were also observed from the surface waters to the deep chlorophyll maximum layer in the open ocean. Only three clusters contained known cyanophage isolates, while the identities of the other six clusters remain unknown. Whether or not these unidentified groups are composed of bacteriophages that infect different Synechococcus groups or other closely related cyanobacteria remains to be determined. The high genetic diversity of marine cyanophage assemblages revealed by the g20 sequences suggests that marine viruses can potentially play important roles in regulating microbial genetic diversity.
Figures
References
-
- Bergh, O., K. Y. Borsheim, G. Bratbak, and M. Heldal. 1989. High abundance of viruses found in aquatic environments. Nature (London) 340:467-468. - PubMed
-
- Chen, F., and C. A. Suttle. 1996. Evolutionary relationships among large double-stranded DNA viruses that infect microalgae and other organisms as inferred from DNA polymerase genes. Virology 219:170-178. - PubMed
-
- Chisholm, S. W., R. J. Olson, E. R. Zettler, R. Goericke, J. B. Waterbury, and N. A. Welschmeyer. 1988. A novel free-living prochlorophyte abundant in the oceanic euphotic zone. Nature (London) 334:340-343.
-
- Felsenstein, J. 1993. PHYLIP: phylogeny inference package (version 3.5). University of Washington, Seattle.
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
