

Role of p75 neurotrophin receptor in the neurotoxicity by beta-amyloid peptides and synergistic effect of inflammatory cytokines
- PMID: 11927634
- PMCID: PMC2193732
- DOI: 10.1084/jem.20011797
Role of p75 neurotrophin receptor in the neurotoxicity by beta-amyloid peptides and synergistic effect of inflammatory cytokines
Erratum in
- J Exp Med 2002 May 6;195(9):1231
Abstract
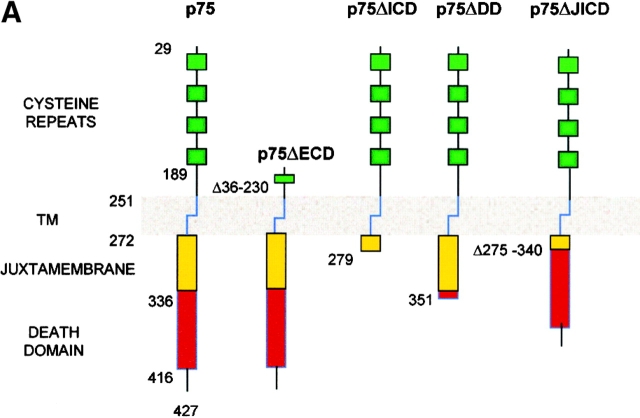
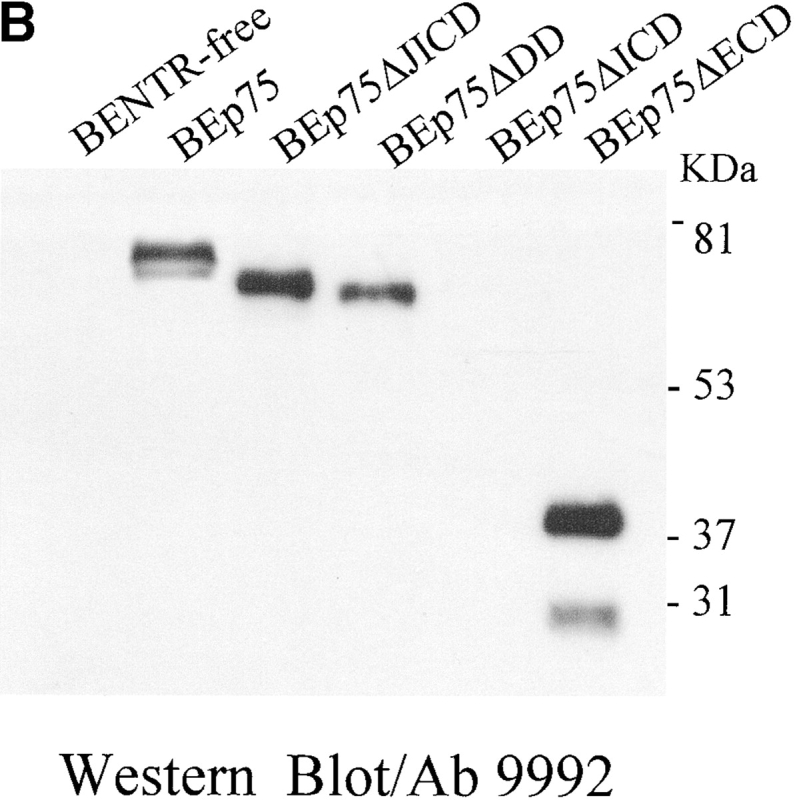
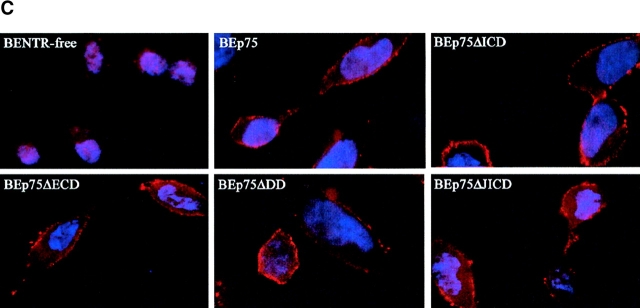
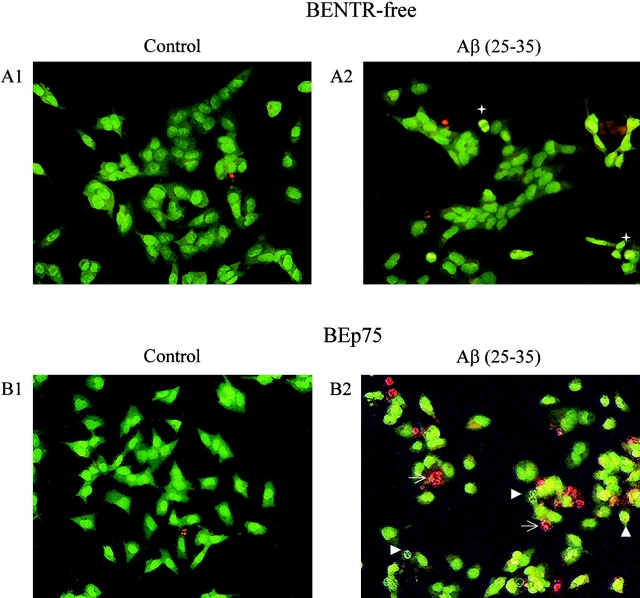
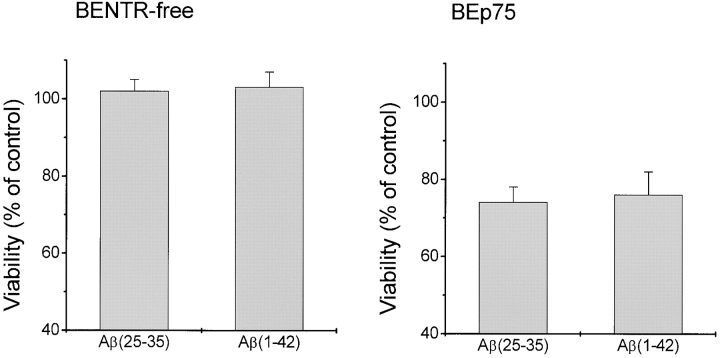
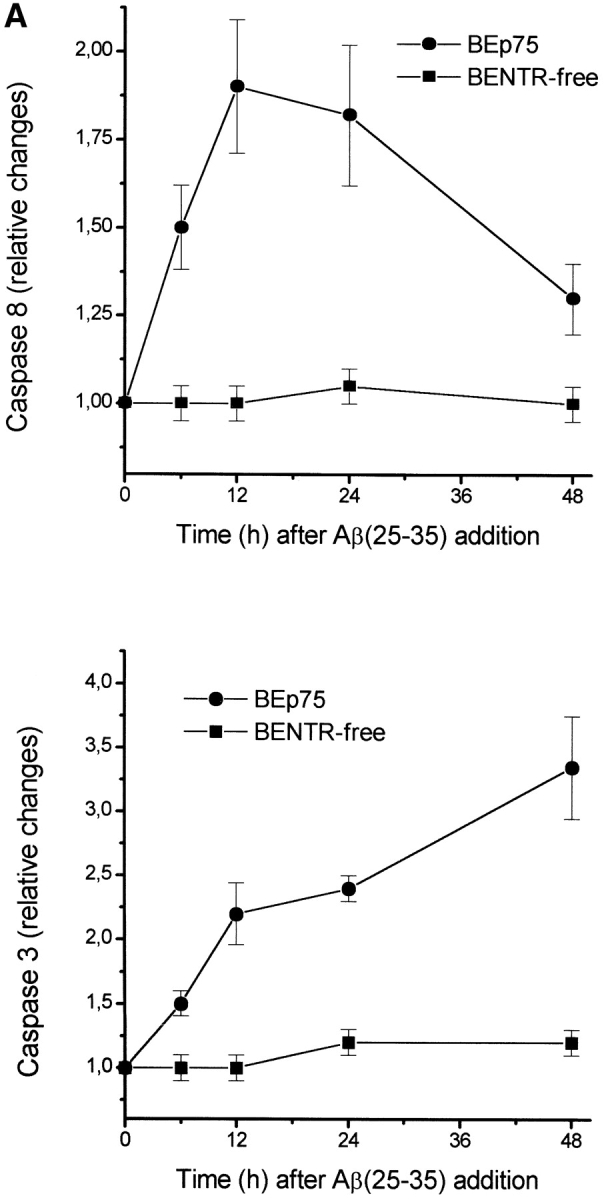
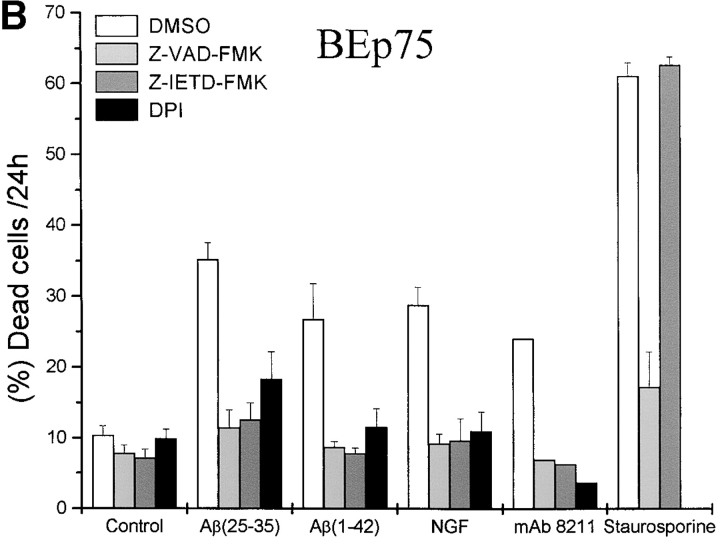
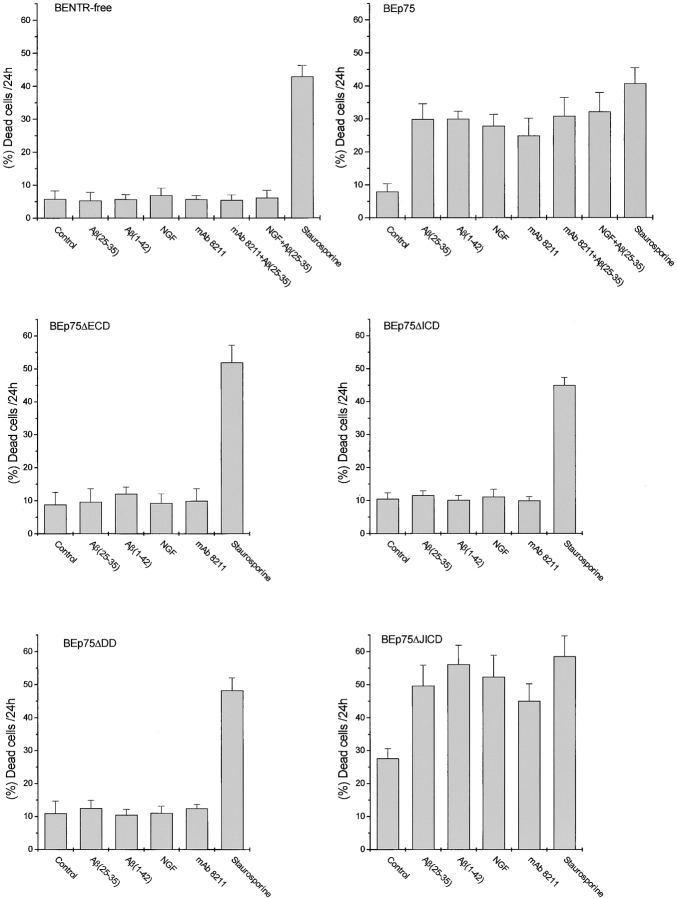
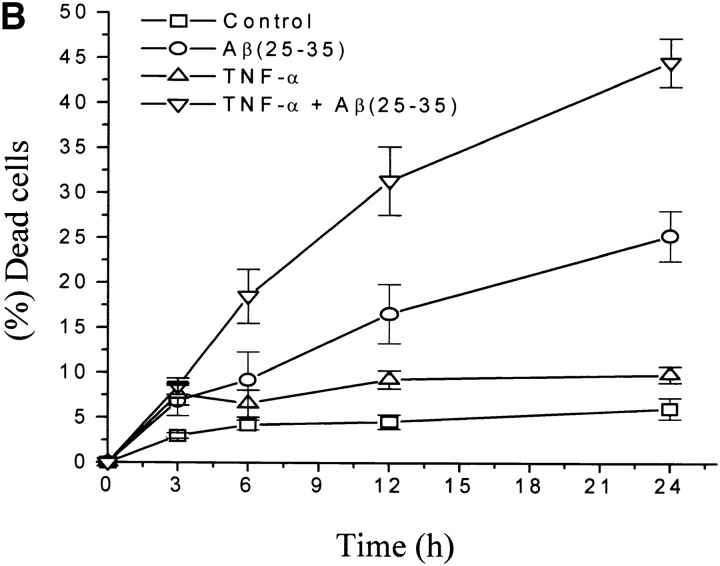
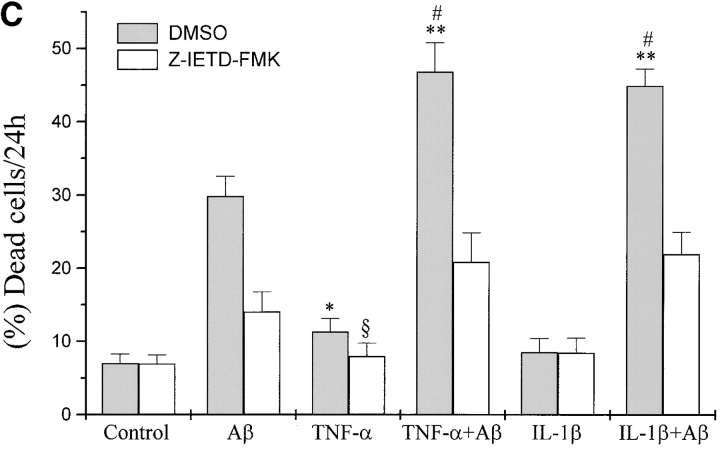
The neurodegenerative changes in Alzheimer's disease (AD) are elicited by the accumulation of beta-amyloid peptides (Abeta), which damage neurons either directly by interacting with components of the cell surface to trigger cell death signaling or indirectly by activating astrocytes and microglia to produce inflammatory mediators. It has been recently proposed that the p75 neurotrophin receptor (p75(NTR)) is responsible for neuronal damage by interacting with Abeta. By using neuroblastoma cell clones lacking the expression of all neurotrophin receptors or engineered to express full-length or various truncated forms of p75(NTR), we could show that p75(NTR) is involved in the direct signaling of cell death by Abeta via the function of its death domain. This signaling leads to the activation of caspases-8 and -3, the production of reactive oxygen intermediates and the induction of an oxidative stress. We also found that the direct and indirect (inflammatory) mechanisms of neuronal damage by Abeta could act synergistically. In fact, TNF-alpha and IL-1beta, cytokines produced by Abeta-activated microglia, could potentiate the neurotoxic action of Abeta mediated by p75(NTR) signaling. Together, our results indicate that neurons expressing p75(NTR), mostly if expressing also proinflammatory cytokine receptors, might be preferential targets of the cytotoxic action of Abeta in AD.
Figures
References
-
- Hardy, J., and D. Allsop. 1991. Amyloid deposition as the central event in the aetiology of Alzheimer's disease. Trends Pharmacol. Sci. 12:383–388. - PubMed
-
- Yankner, B.A., and M.M. Mesulam. 1991. β–amyloid and the pathogenesis of Alzheimer's disease. N. Engl. J. Med. 325:1849–1857. - PubMed
-
- Selkoe, D.J. 1999. Translating cell biology into therapeutic advances in Alzheimer's disease. Nature. 399:A23–A31. - PubMed
-
- Pike, C.J., A.J. Walencewicz, C.G. Glabe, and C.W. Cotman. 1991. In vitro aging of β-amyloid protein causes peptide aggregation and neurotoxicity. Brain Res. 563:311–314. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
