

Structural and functional mutations of the perlecan gene cause Schwartz-Jampel syndrome, with myotonic myopathy and chondrodysplasia
- PMID: 11941538
- PMCID: PMC447613
- DOI: 10.1086/340390
Structural and functional mutations of the perlecan gene cause Schwartz-Jampel syndrome, with myotonic myopathy and chondrodysplasia
Abstract
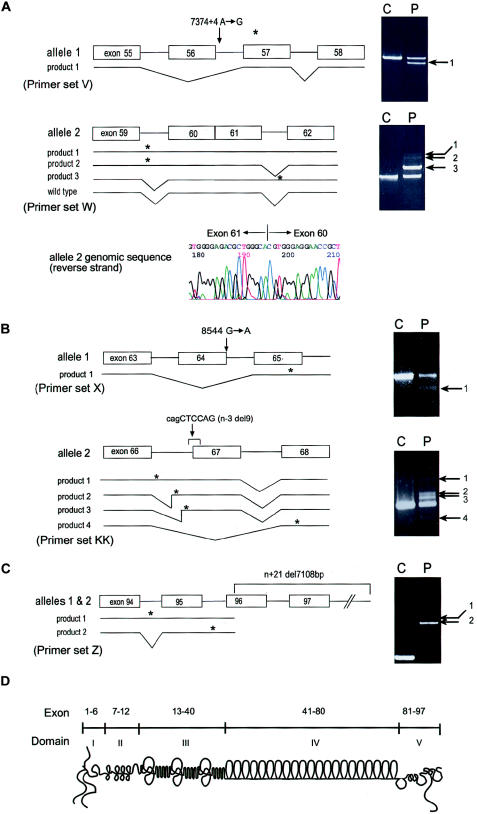
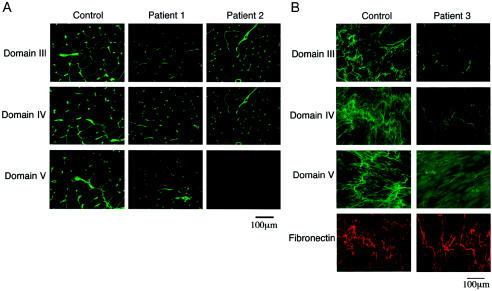
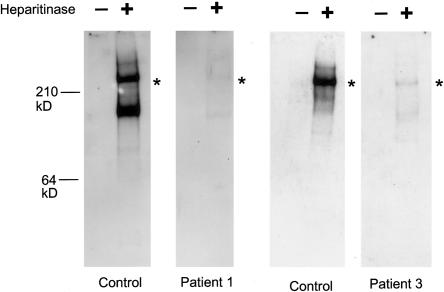
Perlecan, a large heparan sulfate proteoglycan, is a component of the basement membrane and other extracellular matrices and has been implicated in multiple biological functions. Mutations in the perlecan gene (HSPG2) cause two classes of skeletal disorders: the relatively mild Schwartz-Jampel syndrome (SJS) and severe neonatal lethal dyssegmental dysplasia, Silverman-Handmaker type (DDSH). SJS is an autosomal recessive skeletal dysplasia characterized by varying degrees of myotonia and chondrodysplasia, and patients with SJS survive. The molecular mechanism underlying the chondrodystrophic myotonia phenotype of SJS is unknown. In the present report, we identify five different mutations that resulted in various forms of perlecan in three unrelated patients with SJS. Heterozygous mutations in two patients with SJS either produced truncated perlecan that lacked domain V or significantly reduced levels of wild-type perlecan. The third patient had a homozygous 7-kb deletion that resulted in reduced amounts of nearly full-length perlecan. Unlike DDSH, the SJS mutations result in different forms of perlecan in reduced levels that are secreted to the extracellular matrix and are likely partially functional. These findings suggest that perlecan has an important role in neuromuscular function and cartilage formation, and they define the molecular basis involved in the difference in the phenotypic severity between DDSH and SJS.
Figures
References
Electronic-Database Information
-
- GenBank, http://www.ncbi.nlm.nih.gov/GenBank/ (for the sequence of HSPG2 cDNA [accession number M852890] and for HSPG2-containing human chromosome 1 working draft sequence segment [accession number NT_004576])
-
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi..nih.gov/Omin/ (for SJS [MIM 255800] and DDSH [MIM 224410])
References
-
- Aberfeld DC, Hinterbuchner LP, Schneider M (1965) Myotonia, dwarfism, diffuse bone disease and unusual ocular and facial abnormalities (a new syndrome). Brain 88:313–322 - PubMed
-
- Aberfeld DC, Namba T, Vye MV, Grob D (1970) Chondrodystrophic myotonia: report of two cases—myotonia, dwarfism, diffuse bone disease, and unusual ocular and facial abnormalities. Arch Neurol 22:455–462 - PubMed
-
- Arikawa-Hirasawa E, Rossi SG, Rotundo RL, Yamada Y (2002) Absence of acetylcholinesterase at the neuromuscular junctions of perlecan-null mice. Nat Neurosci 5:119–123 - PubMed
-
- Arikawa-Hirasawa E, Watanabe H, Takami H, Hassell JR, Yamada Y (1999) Perlecan is essential for cartilage and cephalic development. Nat Genet 23:354–358 - PubMed
-
- Arikawa-Hirasawa E, Wilcox WR, Le AH, Silverman N, Govindraj P, Hassell JR, Yamada Y (2001) Dyssegmental dysplasia, Silverman-Handmaker type, is caused by functional null mutations of the perlecan gene. Nat Genet 27:431–434 - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
