

Elevated nucleocytoplasmic glycosylation by O-GlcNAc results in insulin resistance associated with defects in Akt activation in 3T3-L1 adipocytes
- PMID: 11959983
- PMCID: PMC122766
- DOI: 10.1073/pnas.072072399
Elevated nucleocytoplasmic glycosylation by O-GlcNAc results in insulin resistance associated with defects in Akt activation in 3T3-L1 adipocytes
Abstract
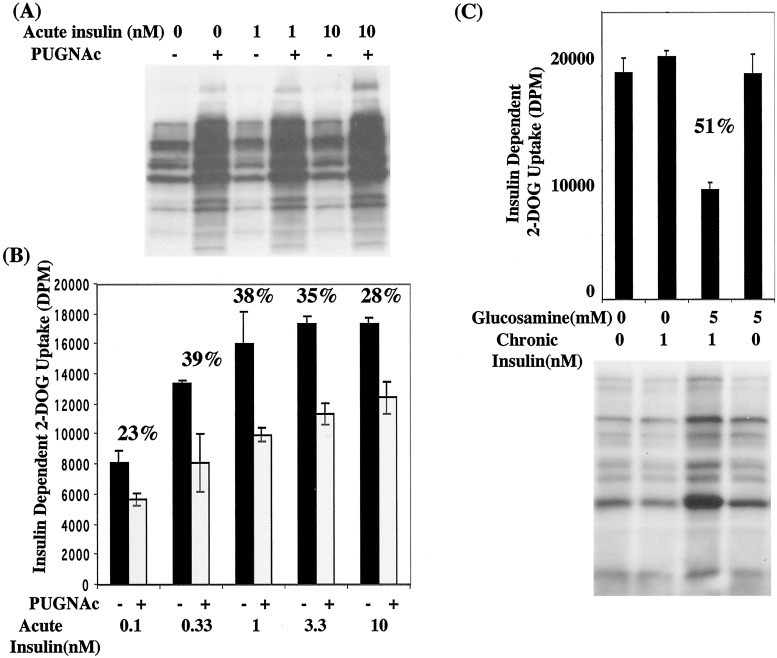
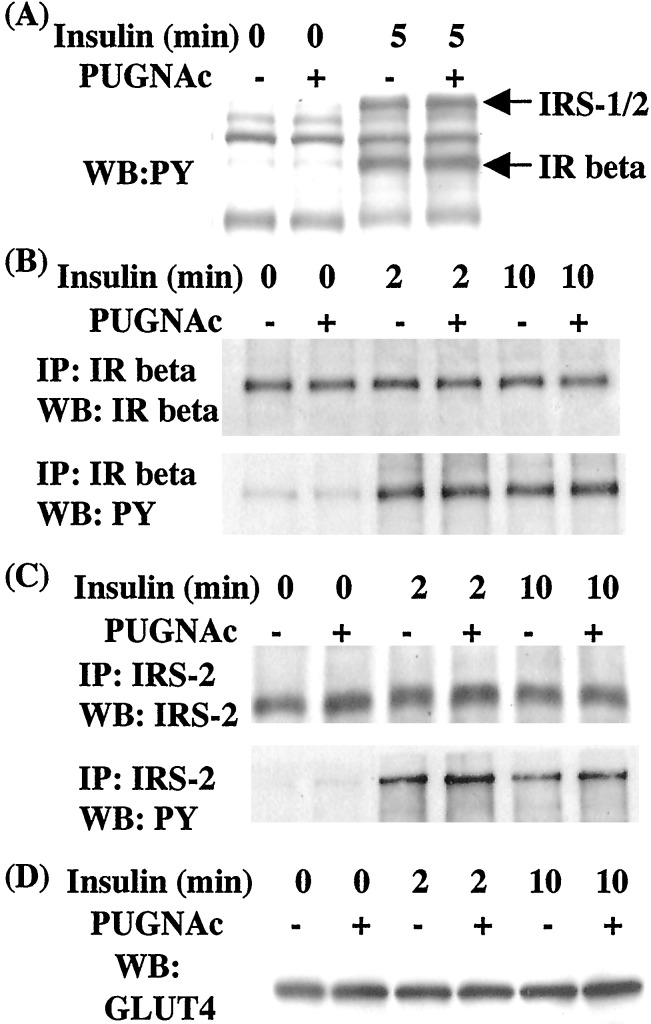
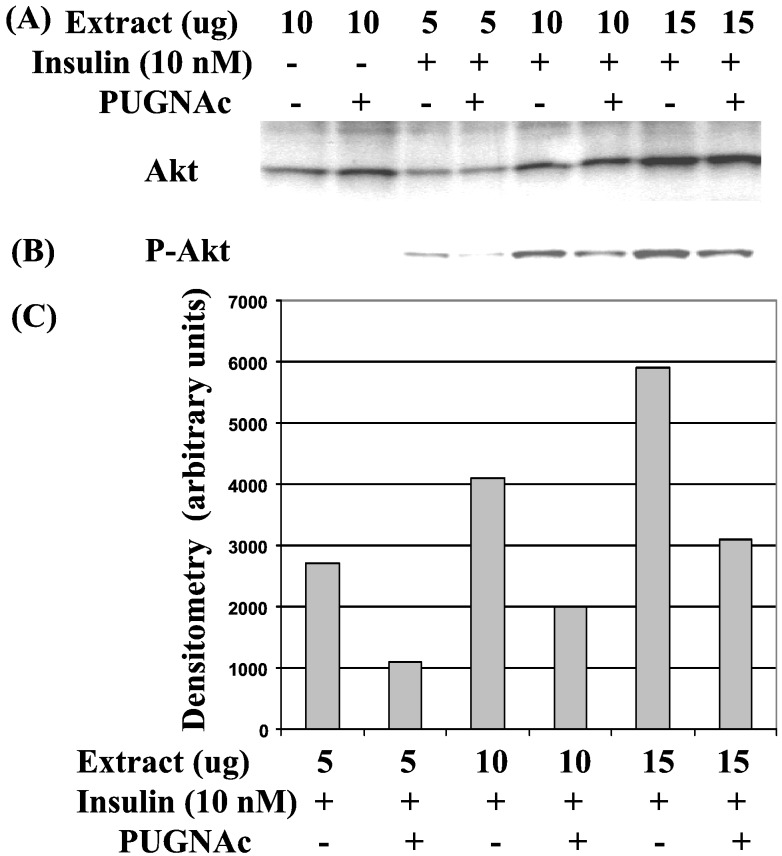
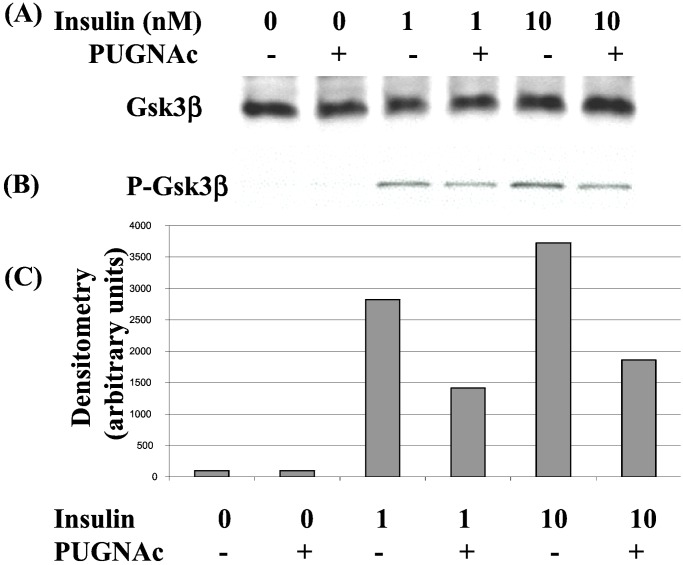
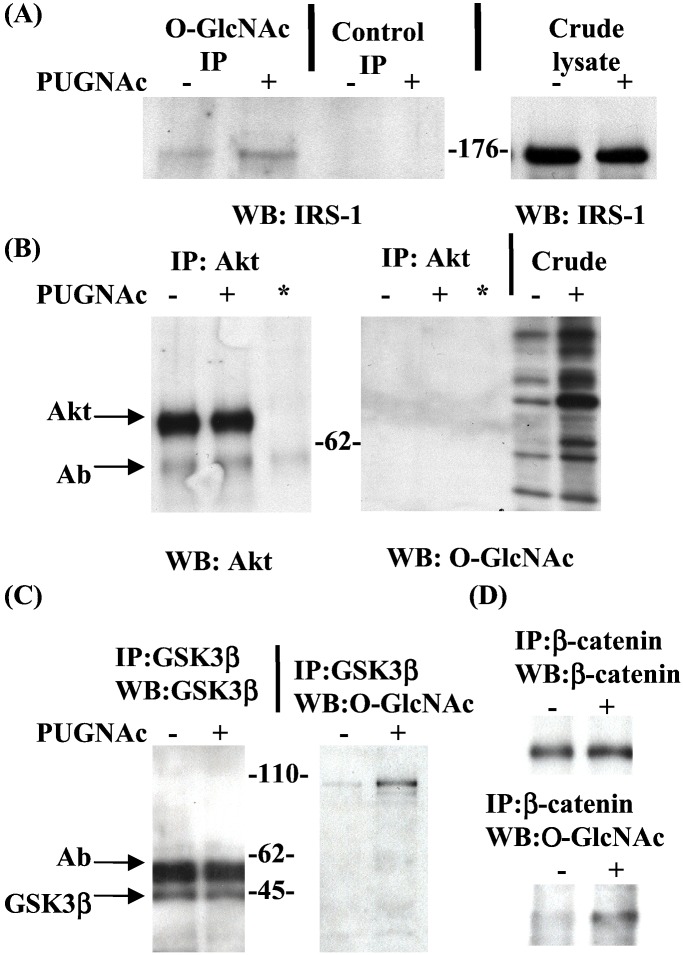
Increased flux of glucose through the hexosamine biosynthetic pathway (HSP) is believed to mediate hyperglycemia-induced insulin resistance in diabetes. The end product of the HSP, UDP beta-N-acetylglucosamine (GlcNAc), is a donor sugar nucleotide for complex glycosylation in the secretory pathway and for O-linked GlcNAc (O-GlcNAc) addition to nucleocytoplasmic proteins. Cycling of the O-GlcNAc posttranslational modification was blocked by pharmacological inhibition of O-GlcNAcase, the enzyme that catalyzes O-GlcNAc removal from proteins, with O-(2-acetamido-2-deoxy-d-glucopyranosylidene)amino-N-phenylcarbamate (PUGNAc). PUGNAc treatment increased levels of O-GlcNAc and caused insulin resistance in 3T3-L1 adipocytes. Insulin resistance induced through the HSP by glucosamine and chronic insulin treatment correlated with increased O-GlcNAc levels on nucleocytoplasmic proteins. Whereas insulin receptor autophosphorylation and insulin receptor substrate 2 tyrosine phosphorylation were not affected by PUGNAc inhibition of O-GlcNAcase, downstream phosphorylation of Akt at Thr-308 and glycogen synthase kinase 3 beta at Ser-9 was inhibited. PUGNAc-induced insulin resistance was associated with increased O-GlcNAc modification of several proteins including insulin receptor substrate 1 and beta-catenin, two important effectors of insulin signaling. These results suggest that elevation of O-GlcNAc levels attenuate insulin signaling and contribute to the mechanism by which increased flux through the HSP leads to insulin resistance in adipocytes.
Figures
References
-
- Yki-Jarvinen H, Koivisto V. Ann Clin Res. 1984;16:74–83. - PubMed
-
- Beck-Nielsen H, Vaag A, Damsbo P, Handberg A, Nielsen O H, Henriksen J E, Thye-Ronn P. Diabetes Care. 1992;15:418–429. - PubMed
-
- Kruszynska Y T, Olefsky J M. J Invest Med. 1996;44:413–428. - PubMed
-
- Hager S R, Jochen A L, Kalkhoff R K. Am J Physiol. 1991;260:E353–E362. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
