

Neuronal deficiency of presenilin 1 inhibits amyloid plaque formation and corrects hippocampal long-term potentiation but not a cognitive defect of amyloid precursor protein [V717I] transgenic mice
- PMID: 11978821
- PMCID: PMC6758348
- DOI: 10.1523/JNEUROSCI.22-09-03445.2002
Neuronal deficiency of presenilin 1 inhibits amyloid plaque formation and corrects hippocampal long-term potentiation but not a cognitive defect of amyloid precursor protein [V717I] transgenic mice
Abstract
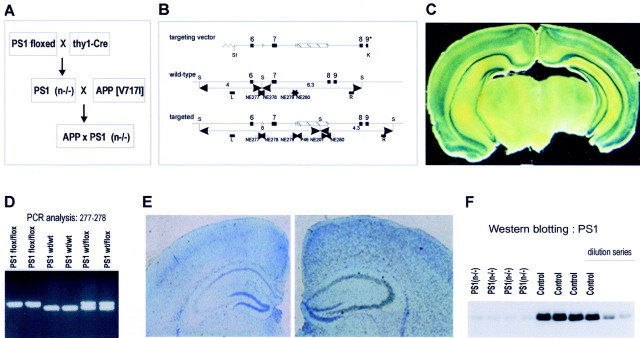
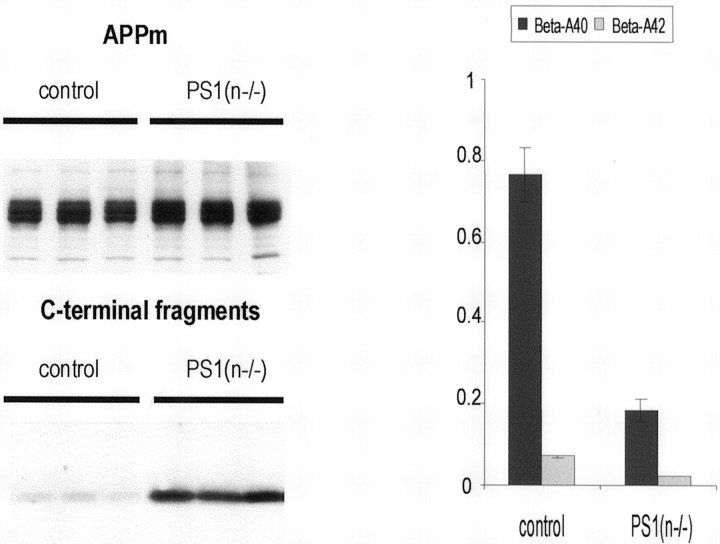
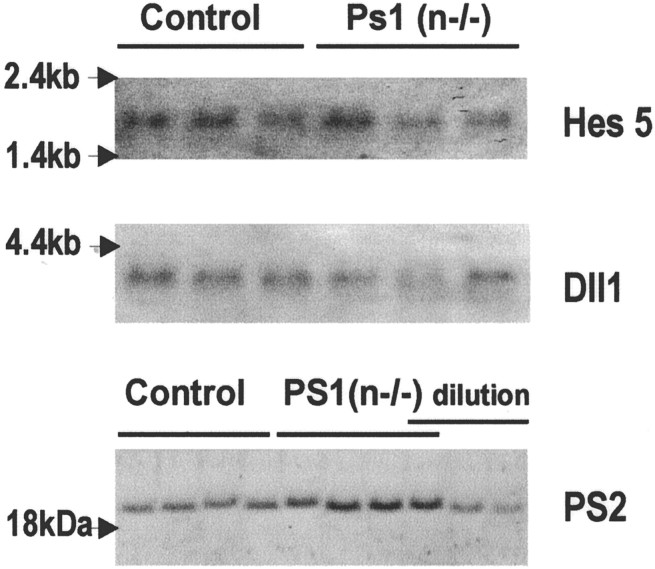
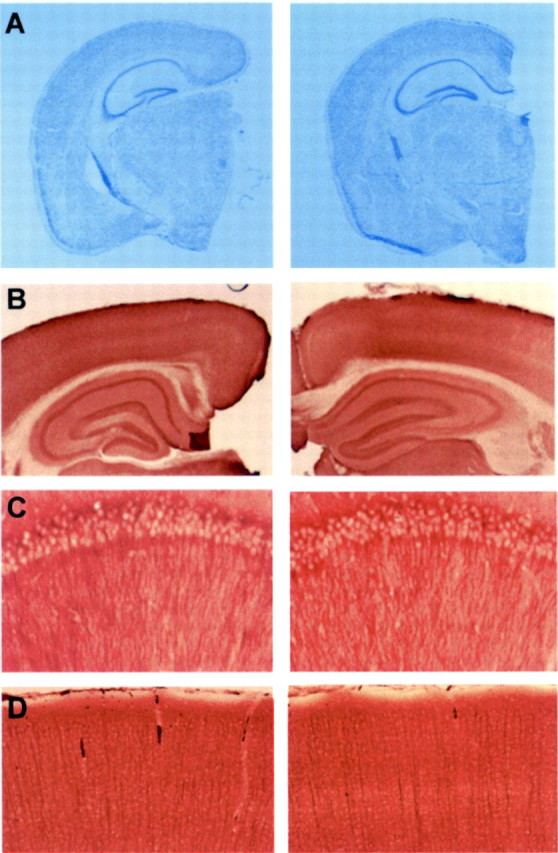
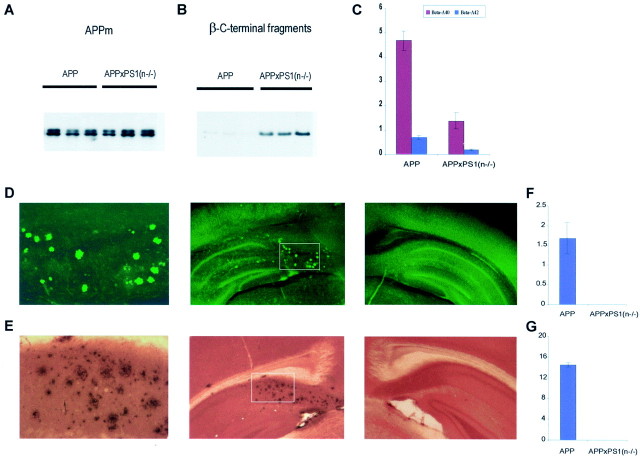
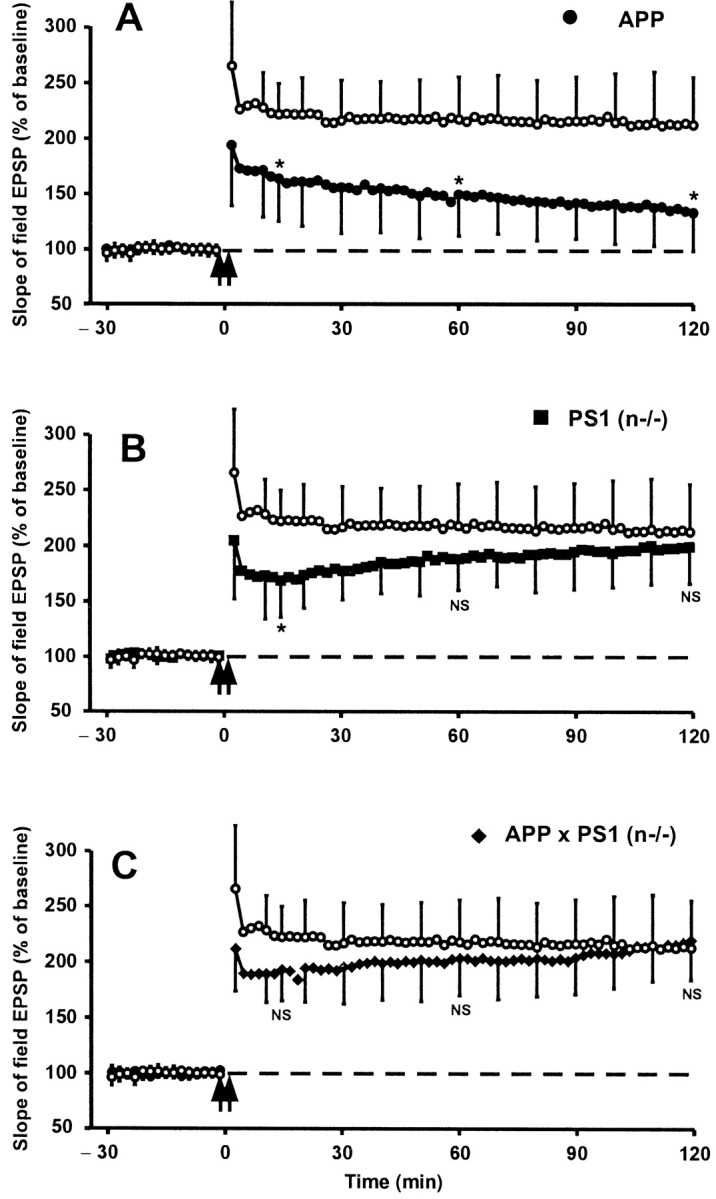
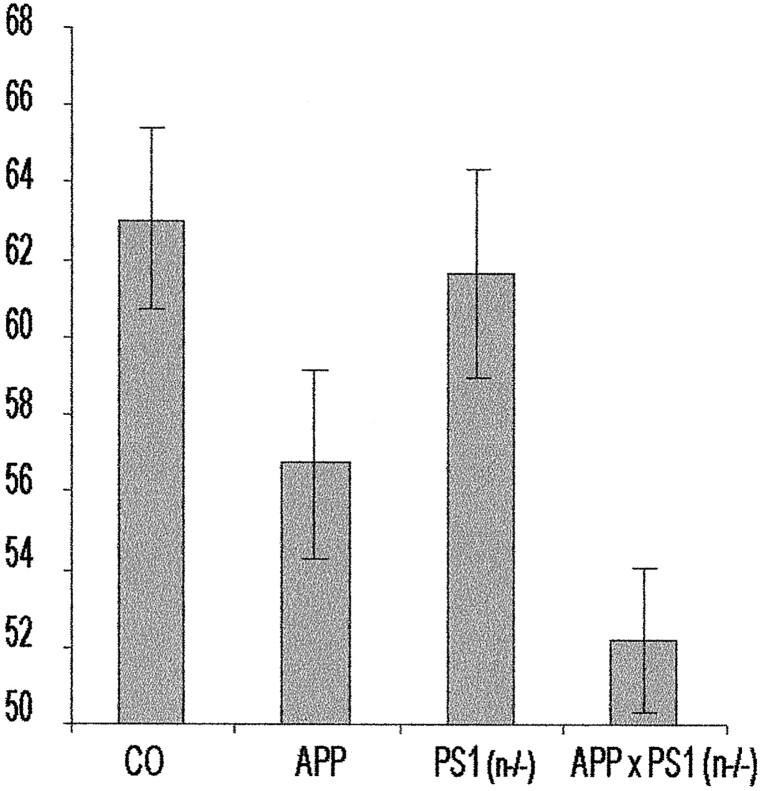
In the brain of Alzheimer's disease (AD) patients, neurotoxic amyloid peptides accumulate and are deposited as senile plaques. A major therapeutic strategy aims to decrease production of amyloid peptides by inhibition of gamma-secretase. Presenilins are polytopic transmembrane proteins that are essential for gamma-secretase activity during development and in amyloid production. By loxP/Cre-recombinase-mediated deletion, we generated mice with postnatal, neuron-specific presenilin-1 (PS1) deficiency, denoted PS1(n-/-), that were viable and fertile, with normal brain morphology. In adult PS1(n-/-) mice, levels of endogenous brain amyloid peptides were strongly decreased, concomitant with accumulation of amyloid precursor protein (APP) C-terminal fragments. In the cross of APP[V717I]xPS1 (n-/-) double transgenic mice, the neuronal absence of PS1 effectively prevented amyloid pathology, even in mice that were 18 months old. This contrasted sharply with APP[V717I] single transgenic mice that all develop amyloid pathology at the age of 10-12 months. In APP[V717I]xPS1 (n-/-) mice, long-term potentiation (LTP) was practically rescued at the end of the 2 hr observation period, again contrasting sharply with the strongly impaired LTP in APP[V717I] mice. The findings demonstrate the critical involvement of amyloid peptides in defective LTP in APP transgenic mice. Although these data open perspectives for therapy of AD by gamma-secretase inhibition, the neuronal absence of PS1 failed to rescue the cognitive defect, assessed by the object recognition test, of the parent APP[V717I] transgenic mice. This points to potentially detrimental effects of accumulating APP C99 fragments and demands further study of the consequences of inhibition of gamma-secretase activity. In addition, our data highlight the complex functional relation of APP and PS1 to cognition and neuronal plasticity in adult and aging brain.
Figures
References
-
- Calhoun ME, Burgermeister P, Phinney AL, Stalder M, Tolnay M, Wiederhold KH, Abramowski D, Sturchler-Pierrat C, Sommer B, Staufenbiel M, Jucker M. Neuronal overexpression of mutant amyloid precursor protein results in prominent deposition of cerebrovascular amyloid. Proc Natl Acad Sci USA. 1999;96:14088–14093. - PMC - PubMed
-
- Chapman PF, White GL, Jones MW, Cooper-Blacketer D, Marshall VJ, Irizarry M, Younkin L, Good MA, Bliss TV, Hyman BT, Younkin SG, Hsiao KK. Impaired synaptic plasticity and learning in aged amyloid precursor protein transgenic mice. Nat Neurosci. 1999;2:271–276. - PubMed
-
- De Strooper B, Saftig P, Craessaerts K, Vanderstichele H, Guhde G, Annaert W, Von Figura K, Van Leuven F. Deficiency of presenilin-1 inhibits the normal cleavage of amyloid precursor protein. Nature. 1998;391:387–390. - PubMed
-
- De Strooper B, Annaert W, Cupers P, Saftig P, Craessaerts K, Mumm JS, Schroeter EH, Schrijvers V, Wolfe MS, Ray WJ, Goate A, Kopan R. A presenilin-1-dependent gamma-secretase-like protease mediates release of Notch intracellular domain. Nature. 1999;398:518–522. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases