

The identification of functional modules from the genomic association of genes
- PMID: 11983890
- PMCID: PMC122872
- DOI: 10.1073/pnas.092632599
The identification of functional modules from the genomic association of genes
Abstract
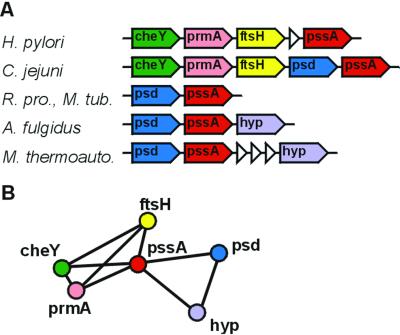
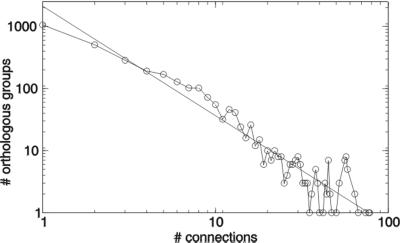
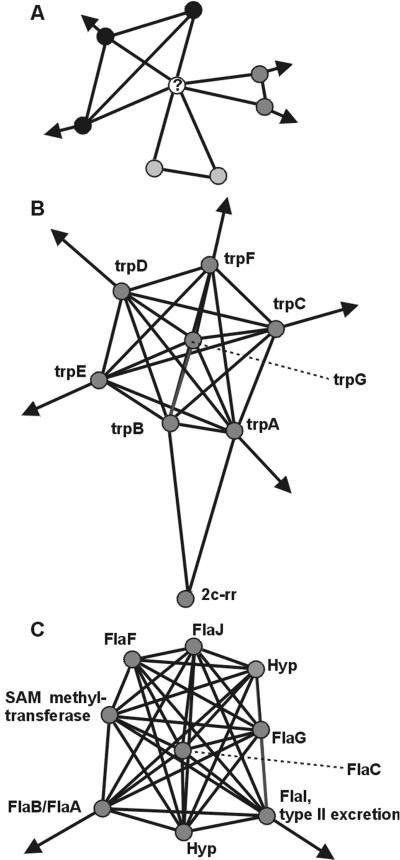
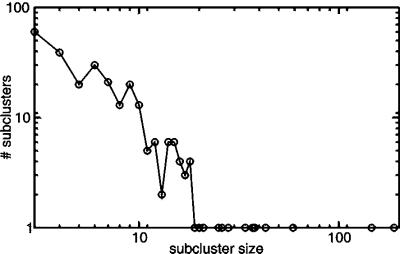
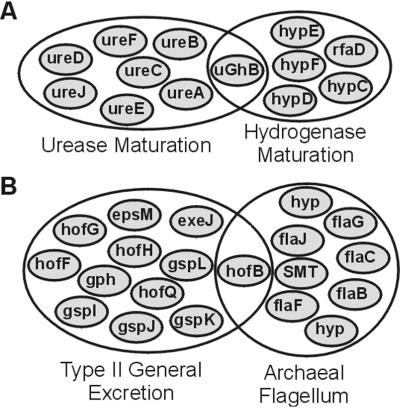
By combining the pairwise interactions between proteins, as predicted by the conserved co-occurrence of their genes in operons, we obtain protein interaction networks. Here we study the properties of such networks to identify functional modules: sets of proteins that together are involved in a biological process. The complete network contains 3,033 orthologous groups of proteins in 38 genomes. It consists of one giant component, containing 1,611 orthologous groups, and of 516 small disjointed clusters that, on average, contain only 2.7 orthologous groups. These small clusters have a homogeneous functional composition and thus represent functional modules in themselves. Analysis of the giant component reveals that it is a scale-free, small-world network with a high degree of local clustering (C = 0.6). It consists of locally highly connected subclusters that are connected to each other by linker proteins. The linker proteins tend to have multiple functions, or are involved in multiple processes and have an above average probability of being essential. By splitting up the giant component at these linker proteins, we identify 265 subclusters that tend to have a homogeneous functional composition. The rare functional inhomogeneities in our subclusters reflect the mixing of different types of (molecular) functions in a single cellular process, exemplified by subclusters containing both metabolic enzymes as well as the transcription factors that regulate them. Comparative genome analysis, thus, allows identification of a level of functional interaction between that of pairwise interactions, and of the complete genome.
Figures
References
-
- Dandekar T, Snel B, Huynen M, Bork P. Trends Biochem Sci. 1998;23:324–328. - PubMed
-
- Overbeek R, Fonstein M, D'Souza M, Pusch G D, Maltsev N. In Silico Biol. 1998;1:0009. http://www.bioinfo.de/isb/1998/01/0009 , http://www.bioinfo.de/isb/1998/01/0009. . - PubMed
-
- Enright A J, Iliopoulos I, Kyrpides N C, Ouzounis C A. Nature (London) 1999;402:86–90. - PubMed
-
- Marcotte E M, Pellegrini M, Ng H L, Rice D W, Yeates T O, Eisenberg D. Science. 1999;285:751–753. - PubMed
MeSH terms
LinkOut - more resources
Full Text Sources
