

Contiguous deletion of the X-linked adrenoleukodystrophy gene (ABCD1) and DXS1357E: a novel neonatal phenotype similar to peroxisomal biogenesis disorders
- PMID: 11992258
- PMCID: PMC419992
- DOI: 10.1086/340849
Contiguous deletion of the X-linked adrenoleukodystrophy gene (ABCD1) and DXS1357E: a novel neonatal phenotype similar to peroxisomal biogenesis disorders
Abstract
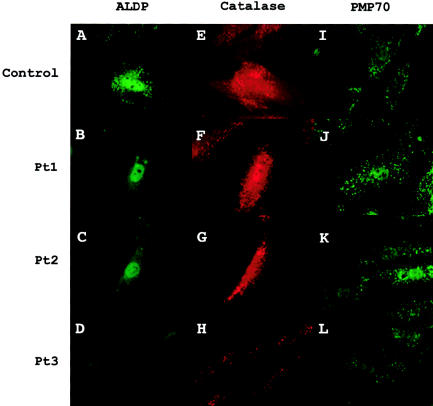
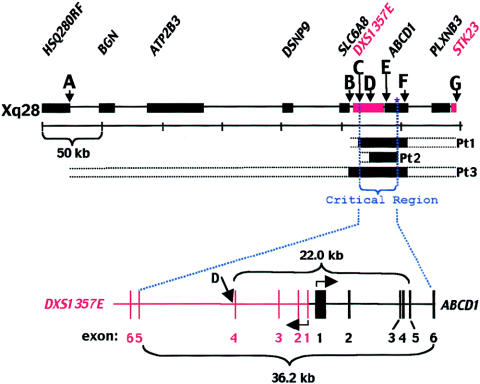
X-linked adrenoleukodystrophy (X-ALD) results from mutations in ABCD1. ABCD1 resides on Xq28 and encodes an integral peroxisomal membrane protein (ALD protein [ALDP]) that is of unknown function and that belongs to the ATP-binding cassette-transporter superfamily. Individuals with ABCD1 mutations accumulate very-long-chain fatty acids (VLCFA) (carbon length >22). Childhood cerebral X-ALD is the most devastating form of the disease. These children have the earliest onset (age 7.2 +/- 1.7 years) among the clinical phenotypes for ABCD1 mutations, but onset does not occur at <3 years of age. Individuals with either peroxisomal biogenesis disorders (PBD) or single-enzyme deficiencies (SED) in the peroxisomal beta-oxidation pathway--disorders such as acyl CoA oxidase deficiency and bifunctional protein deficiency--also accumulate VLCFA, but they present during the neonatal period. Until now, it has been possible to distinguish unequivocally between individuals with these autosomal recessively inherited syndromes and individuals with ABCD1 mutations, on the basis of the clinical presentation and measurement of other biochemical markers. We have identified three newborn boys who had clinical symptoms and initial biochemical results consistent with PBD or SED. In further study, however, we showed that they lacked ALDP, and we identified deletions that extended into the promoter region of ABCD1 and the neighboring gene, DXS1357E. Mutations in DXS1357E and the ABCD1 promoter region have not been described previously. We propose that the term "contiguous ABCD1 DXS1357E deletion syndrome" (CADDS) be used to identify this new contiguous-gene syndrome. The three patients with CADDS who are described here have important implications for genetic counseling, because individuals with CADDS may previously have been misdiagnosed as having an autosomal recessive PBD or SED
Figures

References
Electronic-Database Information
-
- dbSTS: database of “Sequence Tagged Sites,” http://www.ncbi.nlm.nih.gov/dbSTS/ (for markers stSG4965 and stSG39985)
-
- Entrez Nucleotide, http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?db=Nucleotide (for Xq28 DNA sequence surrounding ABCD1, including DXS1357E, SLC6A8, and all other genes shown in [accession number U52111])
-
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/ (for X-ALD [MIM 300100])
-
- X-linked Adrenoleukodystrophy Database, http://www.x-ald.nl
References
-
- Bezman L, Moser AB, Raymond GV, Rinaldo P, Watkins PA, Smith KD, Kass NE, Moser HW (2001) Adrenoleukodystrophy: incidence, new mutation rate, and results of extended family screening. Ann Neurol 49:512–517 - PubMed
-
- Bjorkhem I, Sisfontes L, Bostrom B, Kase BF, Blomstrand R (1986) Simple diagnosis of the Zellweger syndrome by gas-liquid chromatography of dimethylacetals. J Lipid Res 27:786–791 - PubMed
-
- Boehm CD, Cutting GR, Lachtermacher MB, Moser HW, Chong SS (1999) Accurate DNA-based diagnostic and carrier testing for X-linked adrenoleukodystrophy. Mol Genet Metab 66:128–136 - PubMed
-
- Corzo D, Cox G, Hobbs N, Quackenbush EJ, Moser A, Moser H (1999) A new phenotypic variant of X-linked adrenoleukodystrophy (ALD) presenting in the newborn period. Am J Hum Genet Suppl 65:A1295
Publication types
MeSH terms
Substances
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
