

PTPN11 mutations in Noonan syndrome: molecular spectrum, genotype-phenotype correlation, and phenotypic heterogeneity
- PMID: 11992261
- PMCID: PMC379142
- DOI: 10.1086/340847
PTPN11 mutations in Noonan syndrome: molecular spectrum, genotype-phenotype correlation, and phenotypic heterogeneity
Abstract
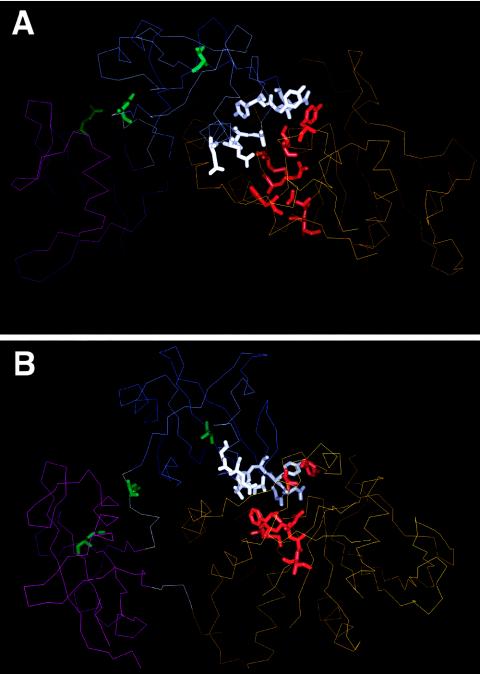
Noonan syndrome (NS) is a developmental disorder characterized by facial dysmorphia, short stature, cardiac defects, and skeletal malformations. We recently demonstrated that mutations in PTPN11, the gene encoding the non-receptor-type protein tyrosine phosphatase SHP-2 (src homology region 2-domain phosphatase-2), cause NS, accounting for approximately 50% of cases of this genetically heterogeneous disorder in a small cohort. All mutations were missense changes and clustered at the interacting portions of the amino-terminal src-homology 2 (N-SH2) and protein tyrosine phosphatase (PTP) domains. A gain of function was postulated as a mechanism for the disease. Here, we report the spectrum and distribution of PTPN11 mutations in a large, well-characterized cohort with NS. Mutations were found in 54 of 119 (45%) unrelated individuals with sporadic or familial NS. There was a significantly higher prevalence of mutations among familial cases than among sporadic ones. All defects were missense, and several were recurrent. The vast majority of mutations altered amino acid residues located in or around the interacting surfaces of the N-SH2 and PTP domains, but defects also affected residues in the C-SH2 domain, as well as in the peptide linking the N-SH2 and C-SH2 domains. Genotype-phenotype analysis revealed that pulmonic stenosis was more prevalent among the group of subjects with NS who had PTPN11 mutations than it was in the group without them (70.6% vs. 46.2%; P<.01), whereas hypertrophic cardiomyopathy was less prevalent among those with PTPN11 mutations (5.9% vs. 26.2%; P<.005). The prevalence of other congenital heart malformations, short stature, pectus deformity, cryptorchidism, and developmental delay did not differ between the two groups. A PTPN11 mutation was identified in a family inheriting Noonan-like/multiple giant-cell lesion syndrome, extending the phenotypic range of disease associated with this gene.
Figures

References
Electronic-Database Information
-
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/ (for NS [MIM 163950] and Noonan-like/multiple giant-cell lesion syndrome [MIM 163955])
References
-
- Arrandale JM, Gore-Willse A, Rock S, Ren J-M, Zhu J, Davis A, Livingston JN, Rabin DU (1996) Insulin signaling in mice expressing reduced levels of Syp. J Biol Chem 271:21353–21358 - PubMed
-
- Bertola DR, Kim CA, Pereira AC, Mota GF, Krieger JE, Vieira IC, Valente M, Loreto MR, Magalhaes RP, Gonzalez CH (2001) Are Noonan syndrome and Noonan-like/multiple giant cell lesion syndrome distinct entities? Am J Med Genet 98:230–234 - PubMed
-
- Bertola DR, Kim CA, Sugayama SMM, Albano LMJ, Wagenfur J, Moyses L, Gonzalez CH (2000) Cardiac findings in 31 patients with Noonan’s syndrome. Arq Bras Cardiol 75:409–412 - PubMed
-
- Brady AF, Jamieson CR, van der Burgt I, Crosby A, van Reen M, Kremer H, Mariman E, Patton MA, Jeffery S (1997) Further delineation of the critical region for Noonan syndrome on the long arm of chromosome 12. Eur J Hum Genet 5:336–337 - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
