

The transcription factor T-bet regulates mucosal T cell activation in experimental colitis and Crohn's disease
- PMID: 11994418
- PMCID: PMC2193714
- DOI: 10.1084/jem.20011956
The transcription factor T-bet regulates mucosal T cell activation in experimental colitis and Crohn's disease
Erratum in
- J Exp Med 2002 Jun 3;195(11):1513
Abstract
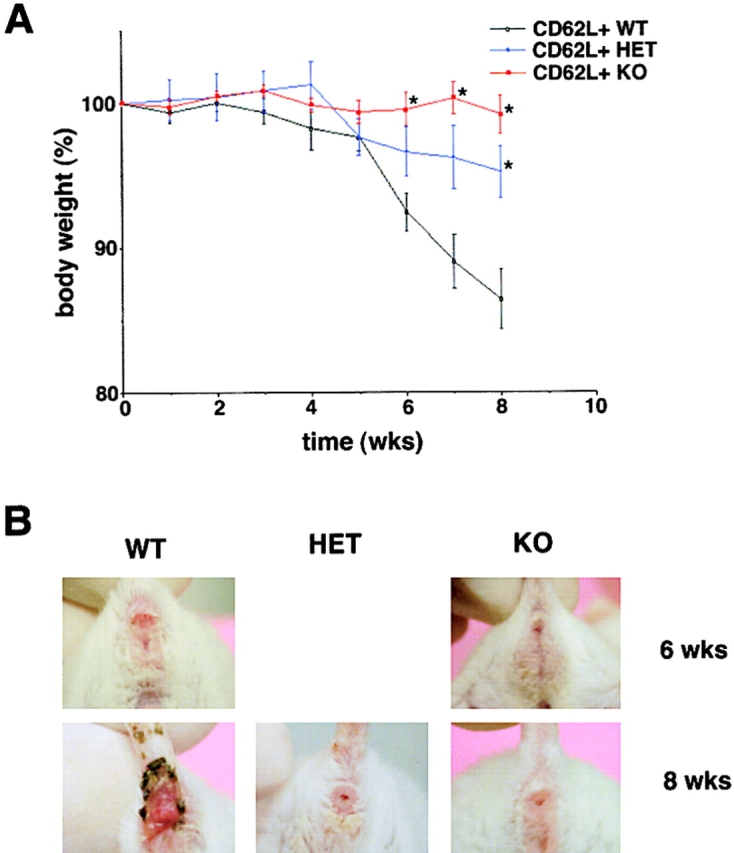
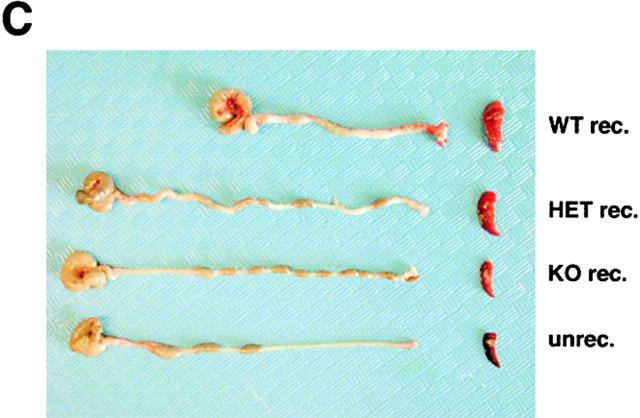
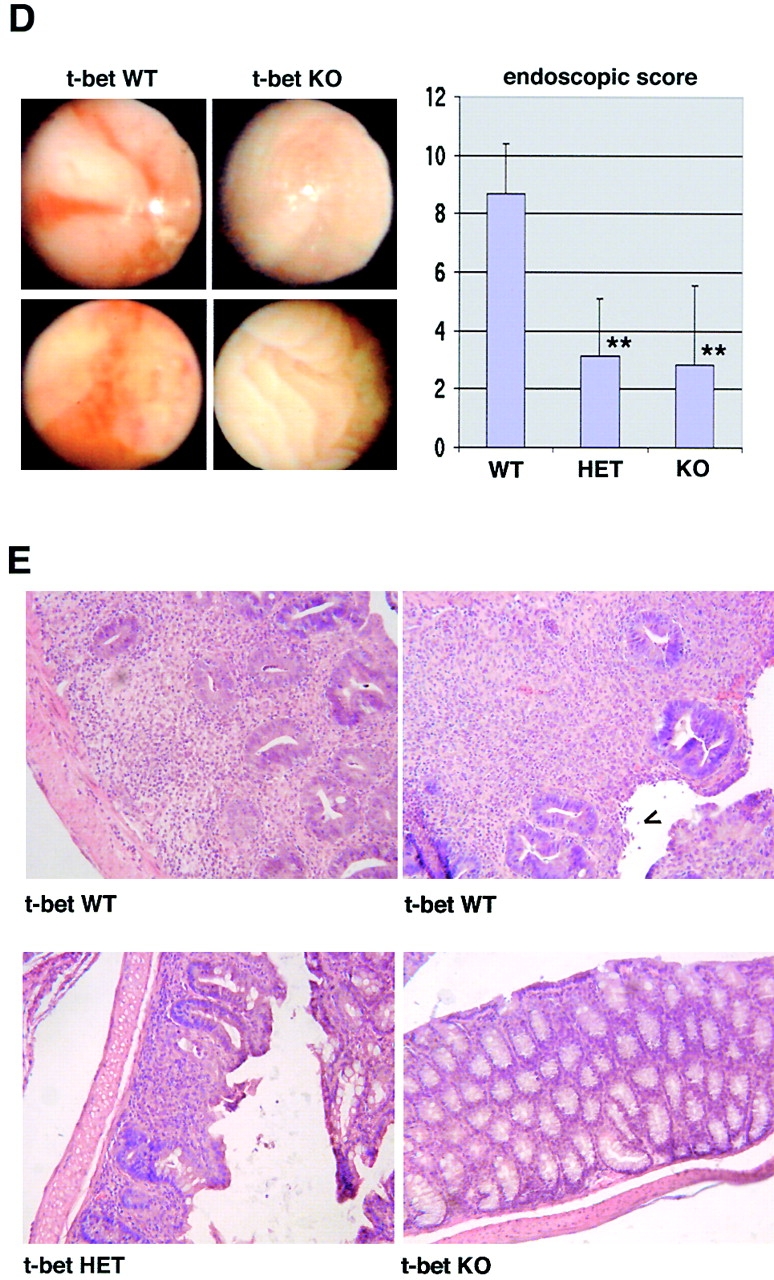
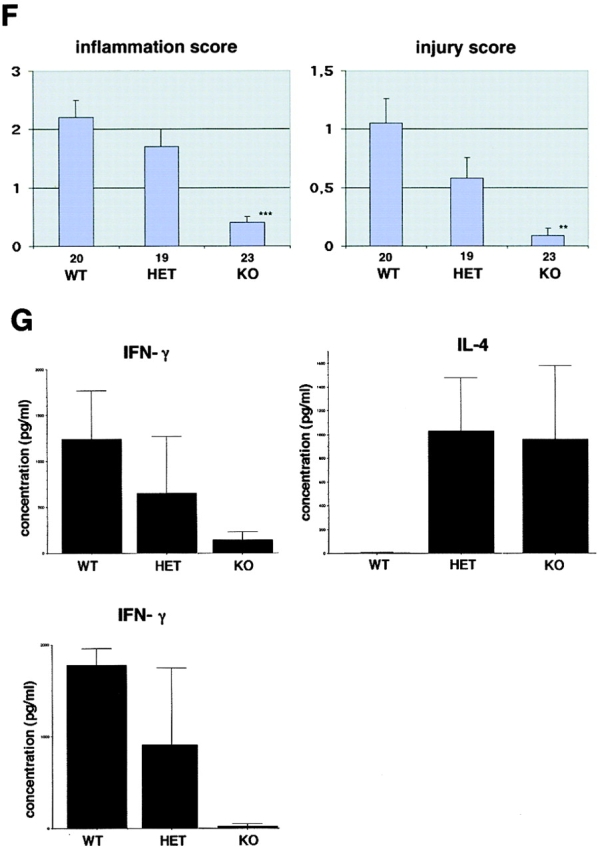
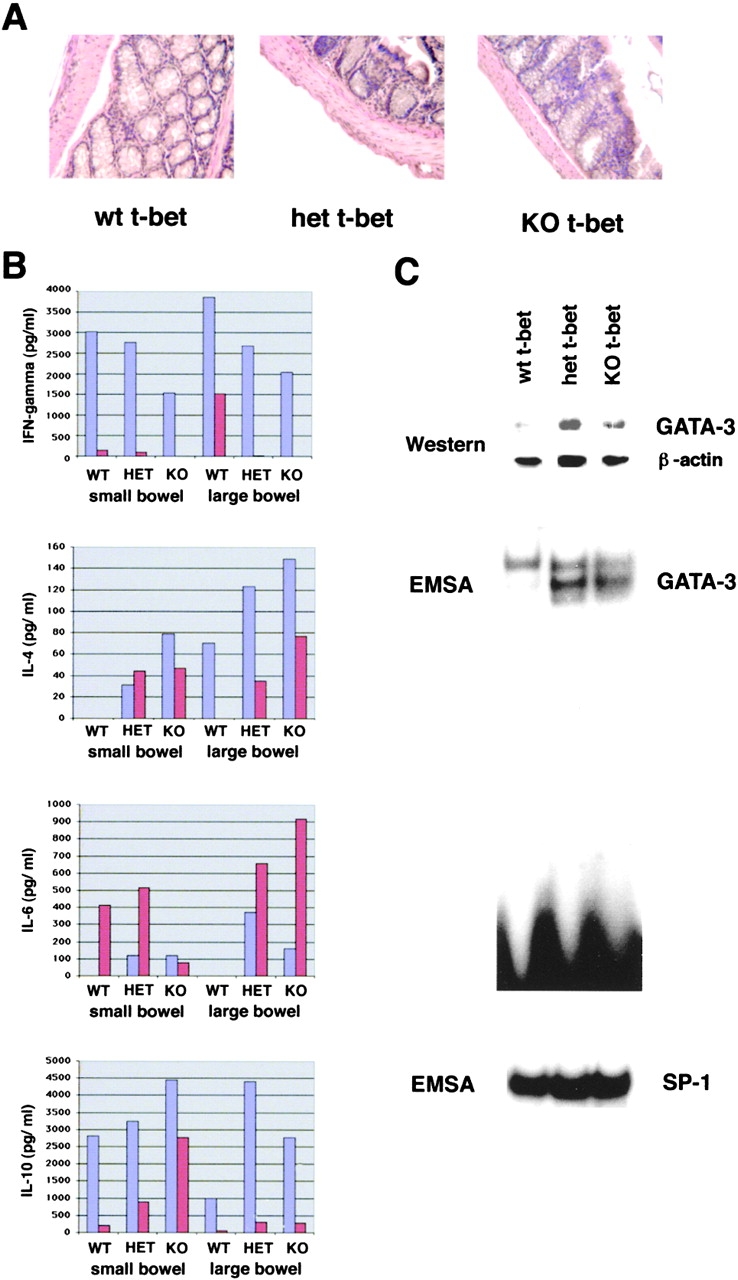
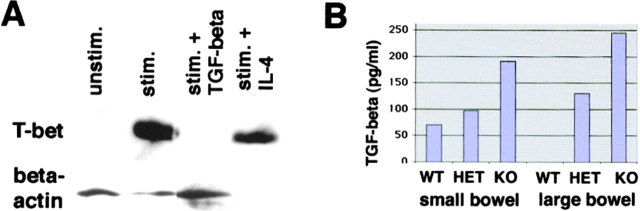
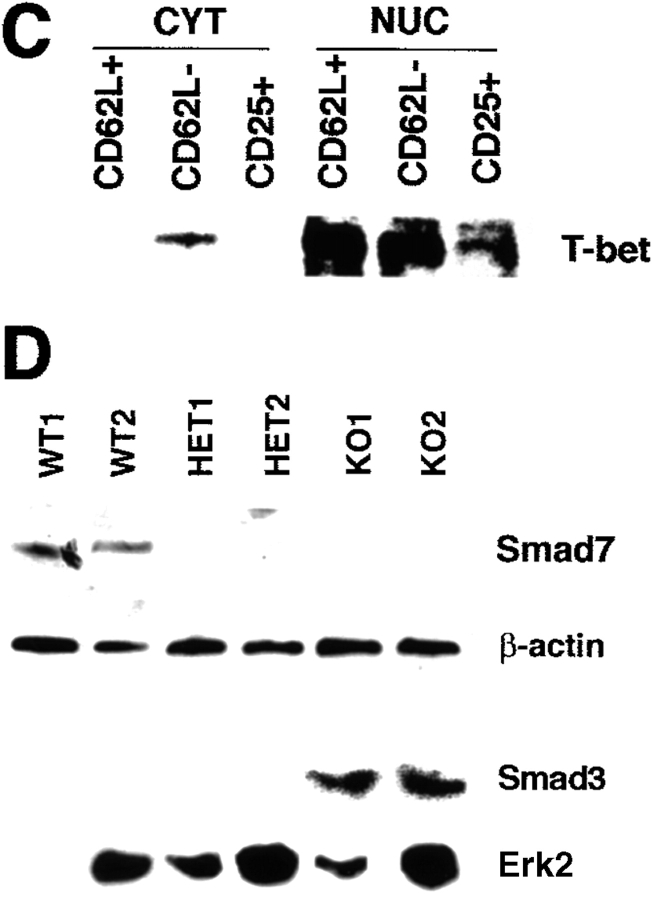
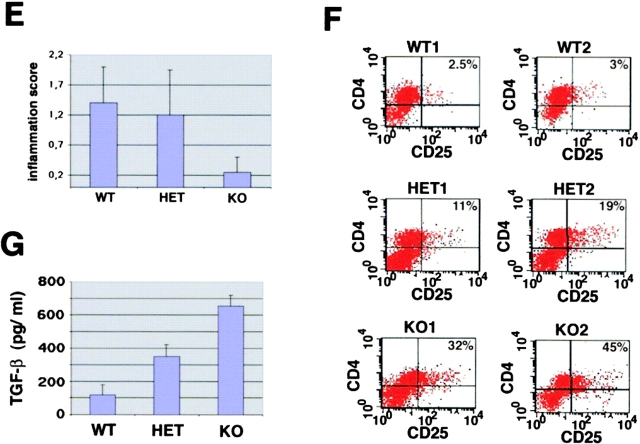
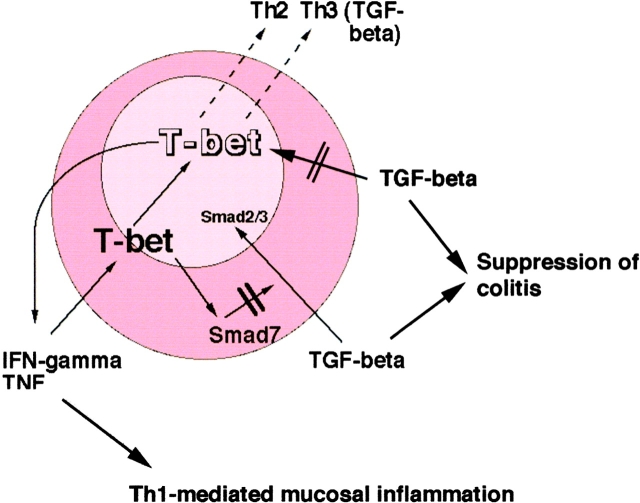
The balance between pro and antiinflammatory cytokines secreted by T cells regulates both the initiation and perpetuation of inflammatory bowel diseases (IBD). In particular, the balance between interferon (IFN)-gamma/interleukin (IL)-4 and transforming growth factor (TGF)-beta activity controls chronic intestinal inflammation. However, the molecular pathways that evoke these responses are not well understood. Here, we describe a critical role for the transcription factor T-bet in controlling the mucosal cytokine balance and clinical disease. We studied the expression and function of T-bet in patients with IBD and in mucosal T cells in various T helper (Th)1- and Th2-mediated animal models of chronic intestinal inflammation by taking advantage of mice that lack T-bet and retroviral transduction techniques, respectively. Whereas retroviral transduction of T-bet in CD62L(+) CD4(+) T cells exacerbated colitis in reconstituted SCID mice, T-bet-deficient T cells failed to induce colitis in adoptive transfer experiments suggesting that overexpression of T-bet is essential and sufficient to promote Th1-mediated colitis in vivo. Furthermore, T-bet-deficient CD62L(-) CD4(+) T cells showed enhanced protective functions in Th1-mediated colitis and exhibited increased TGF-beta signaling suggesting that a T-bet driven pathway of T cell activation controls the intestinal balance between IFN-gamma/IL-4 and TGF-beta responses and the development of chronic intestinal inflammation in T cell-mediated colitis. Furthermore, TGF-beta was found to suppress T-bet expression suggesting a reciprocal relationship between TGF-beta and T-bet in mucosal T cells. In summary, our data suggest a key regulatory role of T-bet in the pathogenesis of T cell-mediated colitis. Specific targeting of this pathway may be a promising novel approach for the treatment of patients with Crohn's disease and other autoimmune diseases mediated by Th1 T lymphocytes.
Figures
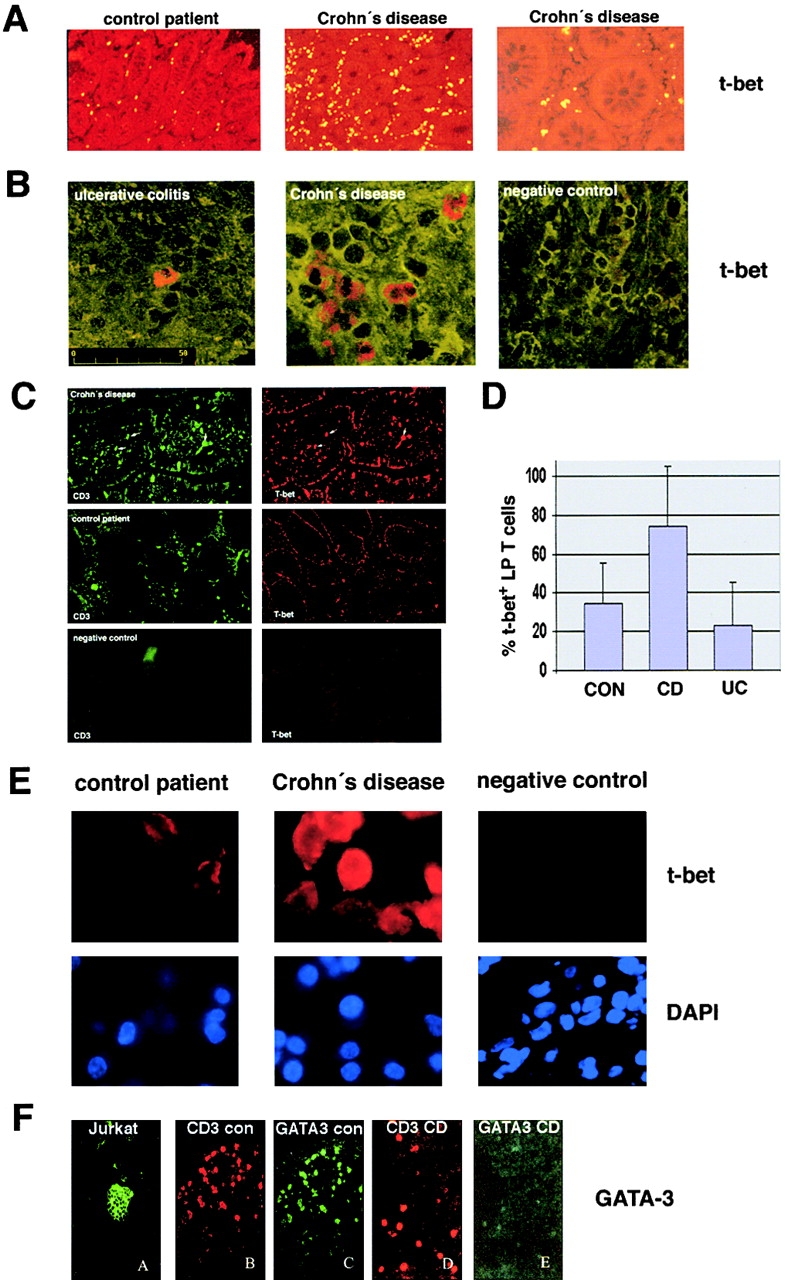
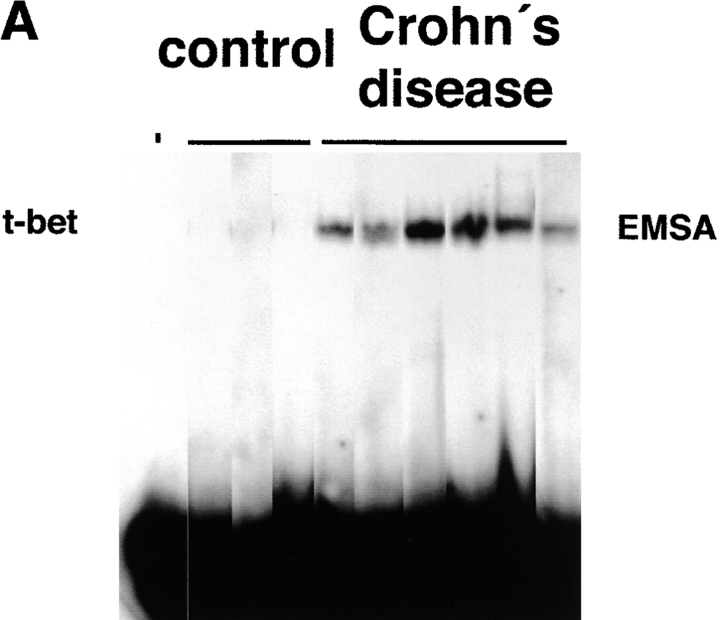
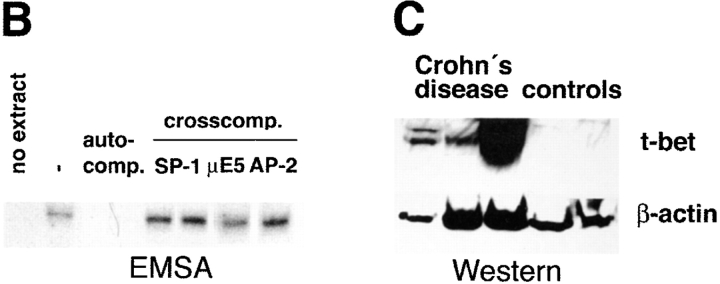
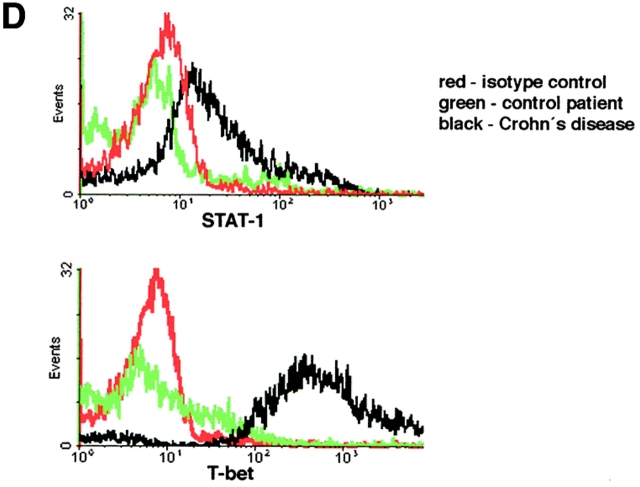
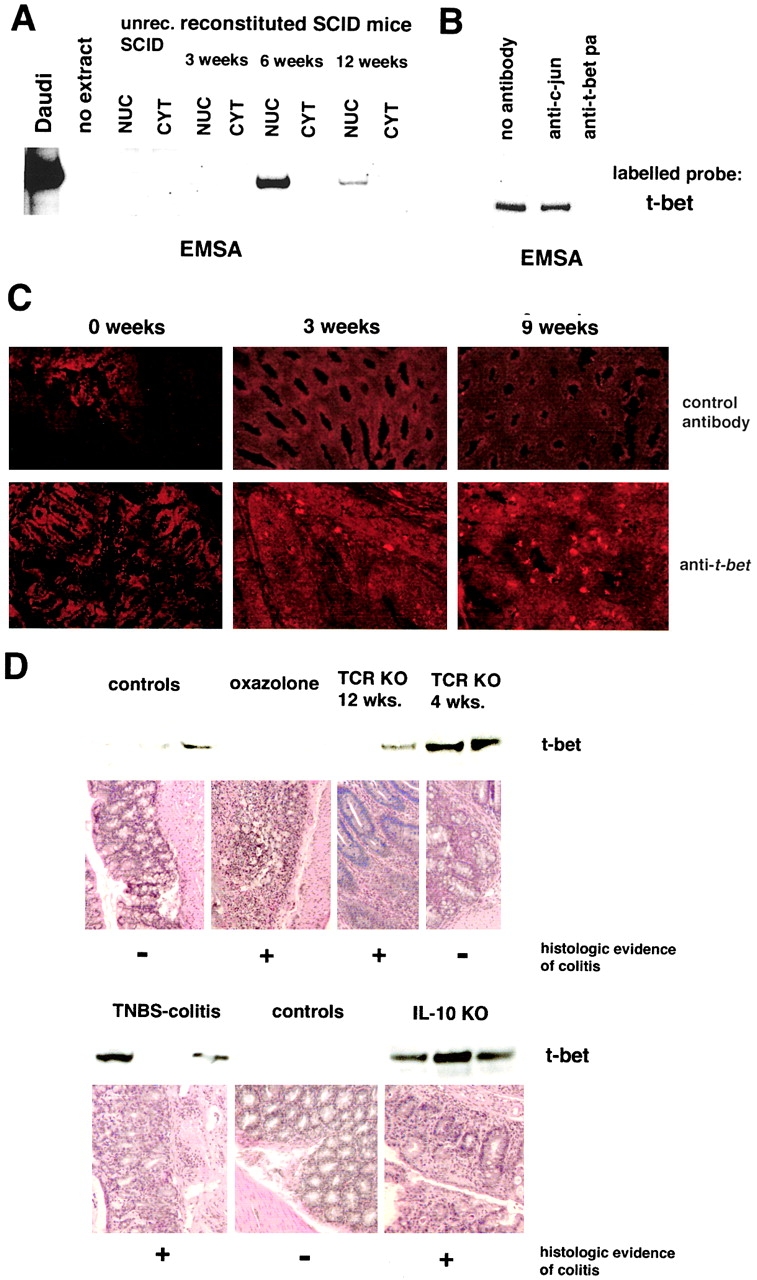
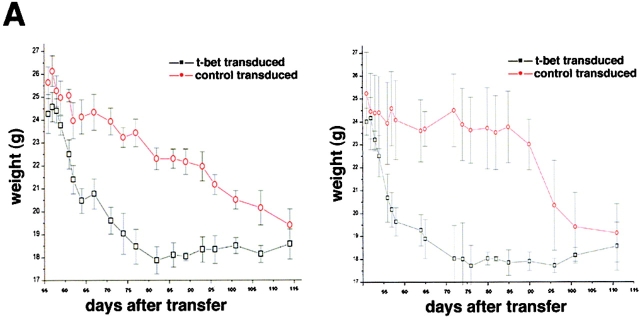
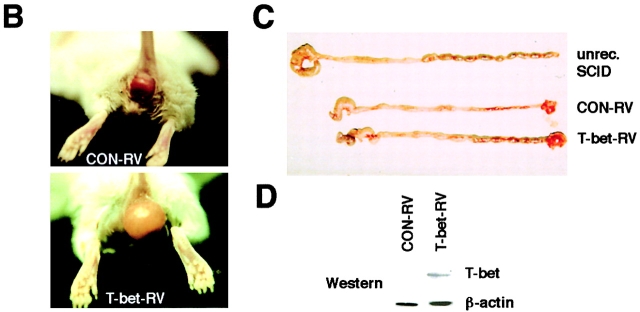

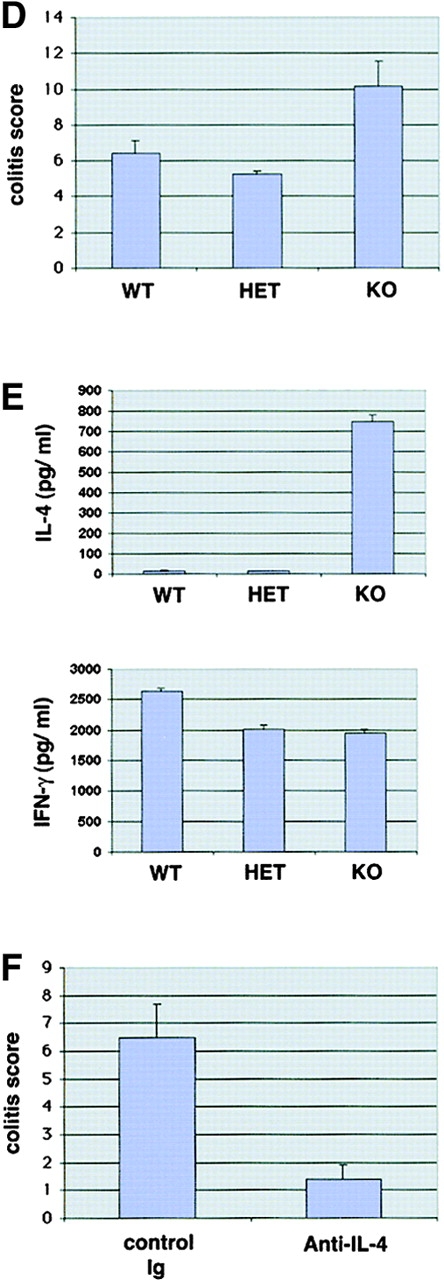
References
-
- Podolsky, D.K. 1991. Inflammatory bowel disease. N. Engl. J. Med. 325:928–937. - PubMed
-
- Neurath, M.F., S. Finotto, and W. Strober. 2001. Immunology of inflammatory bowel disease. Clinical Immunology. 2nd edition. In press. - PubMed
-
- Blumberg, R.S., L.J. Saubermann, and W. Strober. 1999. Animal models of mucosal inflammation and their relation to human inflammatory bowel disease. Curr. Opin. Immunol. 11:648–656. - PubMed
-
- Friedman, S., and R.S. Blumberg. 1999. Inflammatory bowel disease. Harrison's Principles of Internal Medicine. 15th edition. McGraw-Hill, New York. 1679–1691.
-
- Duchmann, R., and M. Zeitz. 1998. Crohn's disease. Handbook of Mucosal Immunology. P. Ogra and W. Strober, editors. Academic Press, NY. 1055–1080.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
