

Inhibitors of COP-mediated transport and cholera toxin action inhibit simian virus 40 infection
- PMID: 12006667
- PMCID: PMC111141
- DOI: 10.1091/mbc.01-12-0592
Inhibitors of COP-mediated transport and cholera toxin action inhibit simian virus 40 infection
Abstract
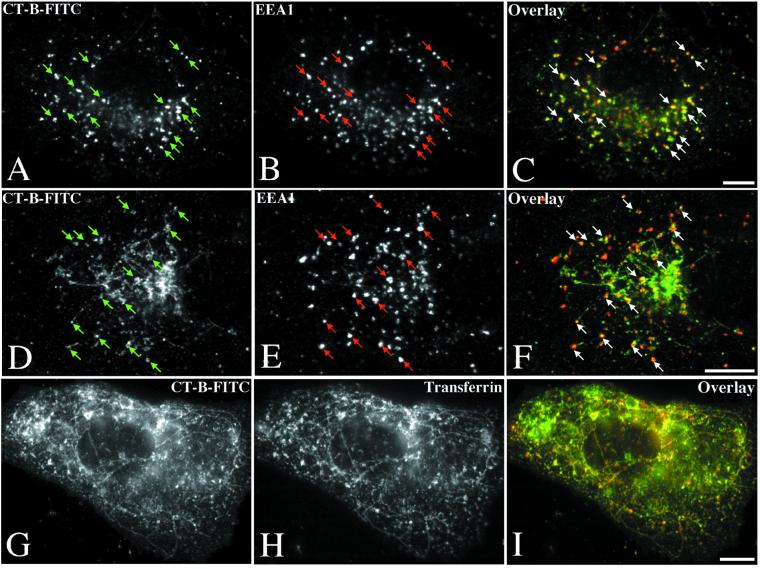
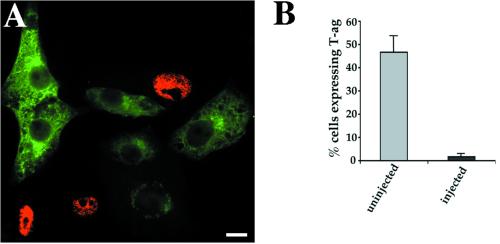
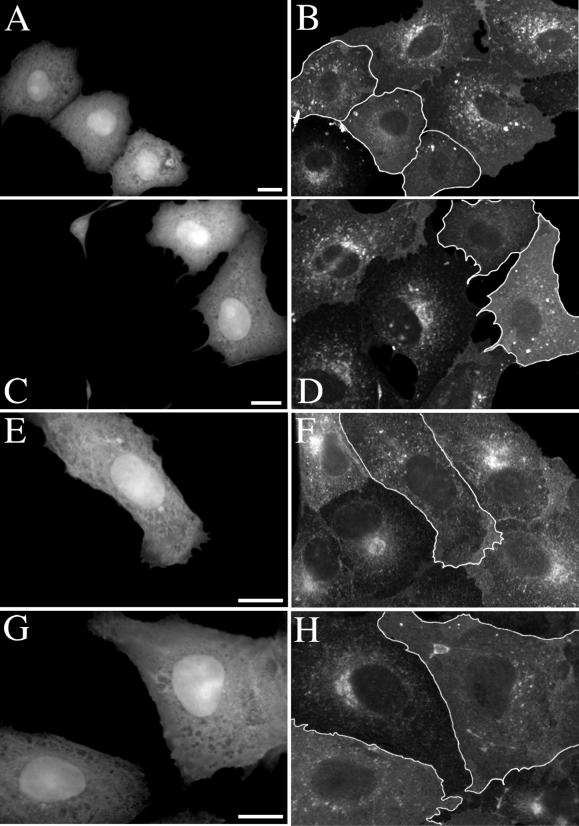
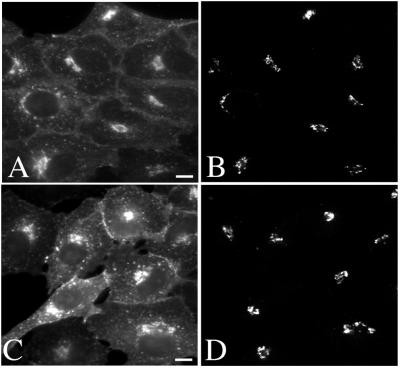
Simian virus 40 (SV40) is a nonenveloped virus that has been shown to pass from surface caveolae to the endoplasmic reticulum in an apparently novel infectious entry pathway. We now show that the initial entry step is blocked by brefeldin A and by incubation at 20 degrees C. Subsequent to the entry step, the virus reaches a domain of the rough endoplasmic reticulum by an unknown pathway. This intracellular trafficking pathway is also brefeldin A sensitive. Infection is strongly inhibited by expression of GTP-restricted ADP-ribosylation factor 1 (Arf1) and Sar1 mutants and by microinjection of antibodies to betaCOP. In addition, we demonstrate a potent inhibition of SV40 infection by the dipeptide N-benzoyl-oxycarbonyl-Gly-Phe-amide, which also inhibits late events in cholera toxin action. Our results identify novel inhibitors of SV40 infection and show that SV40 requires COPI- and COPII-dependent transport steps for successful infection.
Figures







References
-
- Anderson HA, Chen Y, Norkin LC. MHC class I molecules are enriched in caveolae but do not enter with simian virus 40. J Gen Virol. 1998;79:1469–1477. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
