

Sickle cell anaemia: progress in pathogenesis and treatment
- PMID: 12010077
- PMCID: PMC7101942
- DOI: 10.2165/00003495-200262080-00003
Sickle cell anaemia: progress in pathogenesis and treatment
Abstract
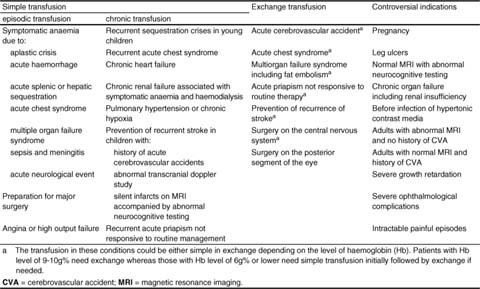
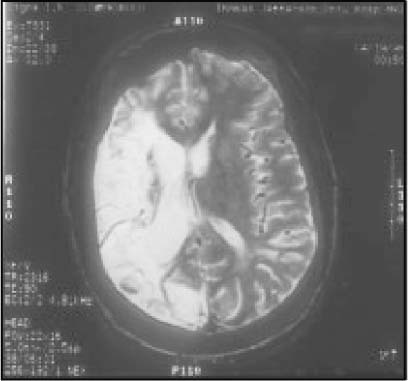
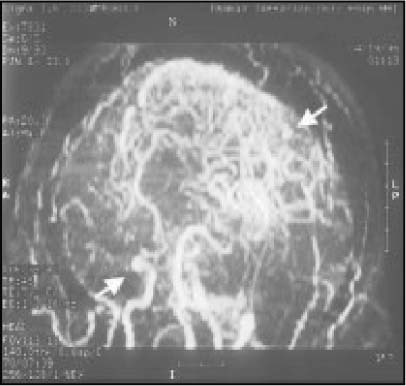
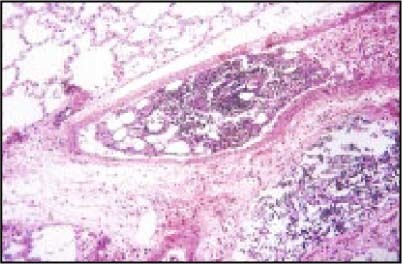
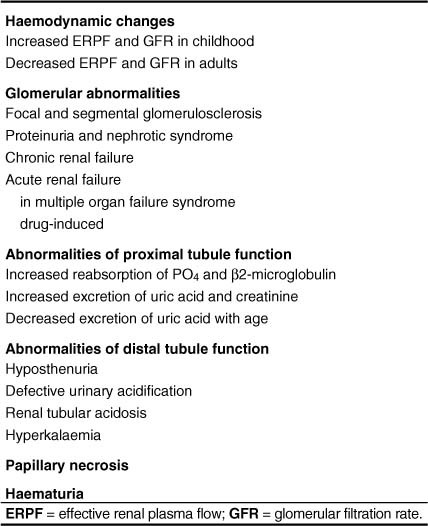
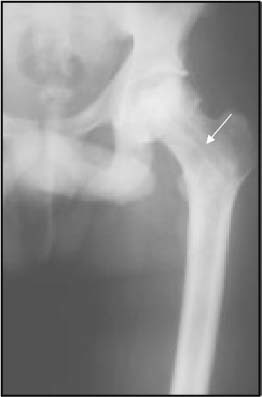
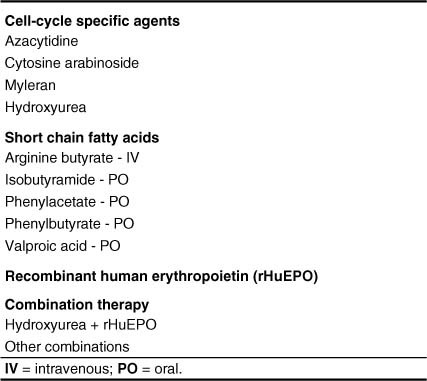
The phenotypic expression of sickle cell anaemia varies greatly among patients and longitudinally in the same patient. It influences all aspects of the life of affected individuals including social interactions, intimate relationships, family relations, peer interactions, education, employment, spirituality and religiosity. The clinical manifestations of sickle cell anaemia are protean and fall into three major categories: anaemia and its sequelae;pain and related issues; andorgan failure including infection. Recent studies on the pathogenesis of sickle cell anaemia have centred on the sequence of events that occur between polymerisation of deoxy haemoglobin (Hb) S and vaso-occlusion. Cellular dehydration, inflammatory response and reperfusion injury seem to be important pathophysiological mechanisms. Management of sickle cell anaemia continues to be primarily palliative in nature, including supportive, symptomatic and preventative approaches to therapy. Empowerment and education are the major aspects of supportive care. Symptomatic management includes pain management, blood transfusion and treatment of organ failure. Pain managment should follow certain priniciples that include assessment, individualisation of therapy and proper utilisation of opioid and nonopioid analgesics in order to acheive adequate pain relief. Blood selected for transfusion should be leuko-reduced and phenotypically matched for the C, E and Kell antigens. Exchange transfusion is indicated in patients who are transfused chronically in order to prevent or delay the onset of iron-overload. Acute chest syndrome is the most common form of organ failure and its management should be agressive, including adequate ventilation, multiple antibacterials and simple or exchange blood transfusion depending on its severity. Preventitive therapy includes prophylactic penicillin in infants and children, blood transfusion (preferably exchange transfusion) in patients with stroke, and hydroxyurea in patients with frequent acute painful episodes. Bone marrow and cord blood transplantation have been successful modalities of curative therapy in selected children with sickle cell anaemia. Newer approaches to preventative therapy include cellular rehydration with agents that inhibit the Gardos channel or the KCl co-transport channel. Curative gene therapy continues to be investigational at the level of the test tube and transgenic mouse models.
Figures











References
-
- Charache S, Dover GJ, Moore RD, et al. Hydroxyurea: effects on hemoglobin F production in patients with sickle cell anemia. Blood. 1992;79:2555–65. - PubMed
-
- Steinberg MH, Barton F, Castro O, et al. Hydroxyurea (HU) is associated with reduced mortality in adults with sickle cell anemia [abstract no. 2087] Blood. 2000;96:485a.
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
