

Emergence of resistant human immunodeficiency virus type 1 in patients receiving fusion inhibitor (T-20) monotherapy
- PMID: 12019106
- PMCID: PMC127242
- DOI: 10.1128/AAC.46.6.1896-1905.2002
Emergence of resistant human immunodeficiency virus type 1 in patients receiving fusion inhibitor (T-20) monotherapy
Abstract
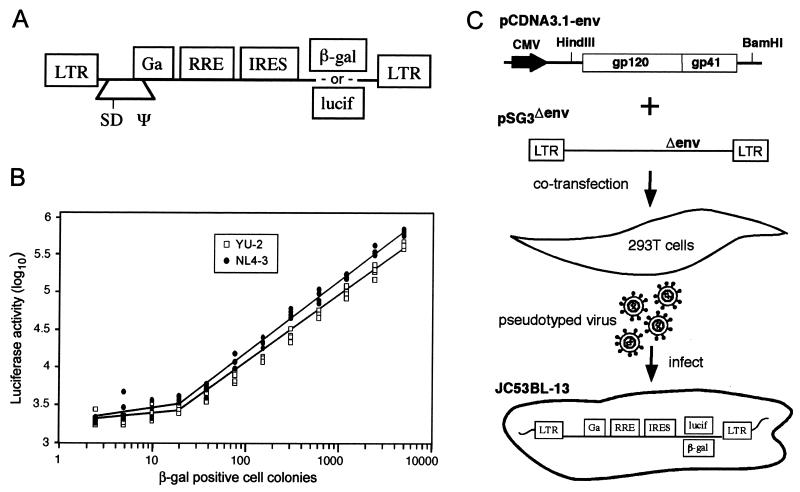
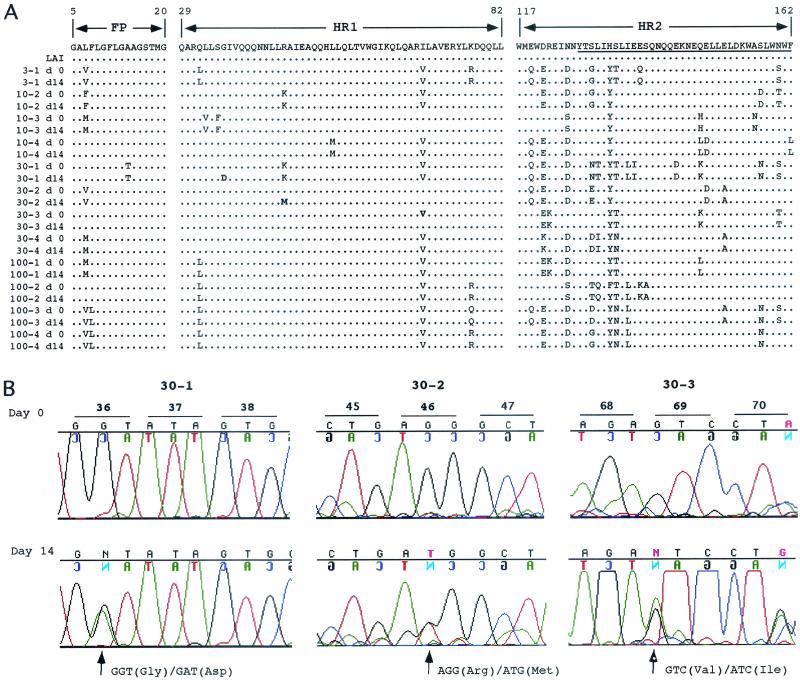
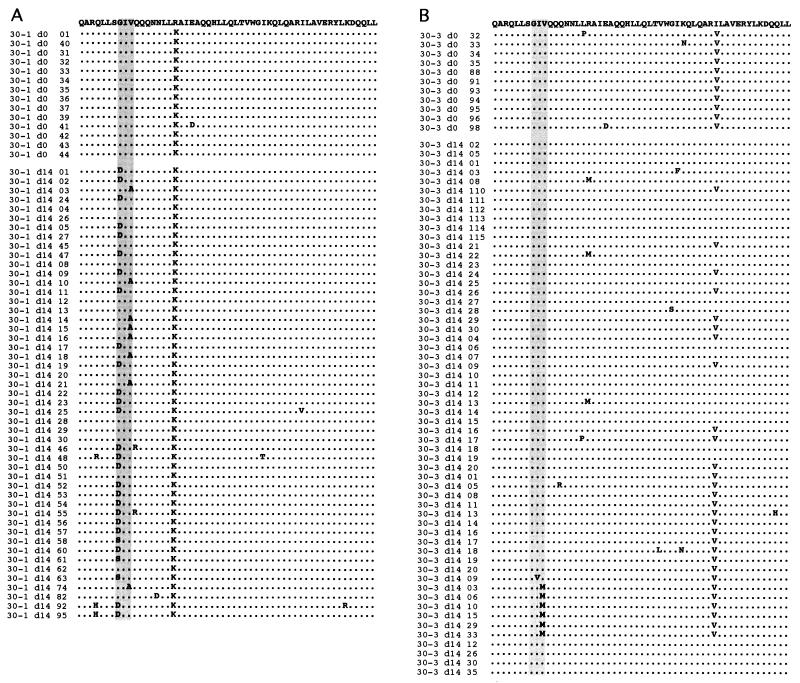
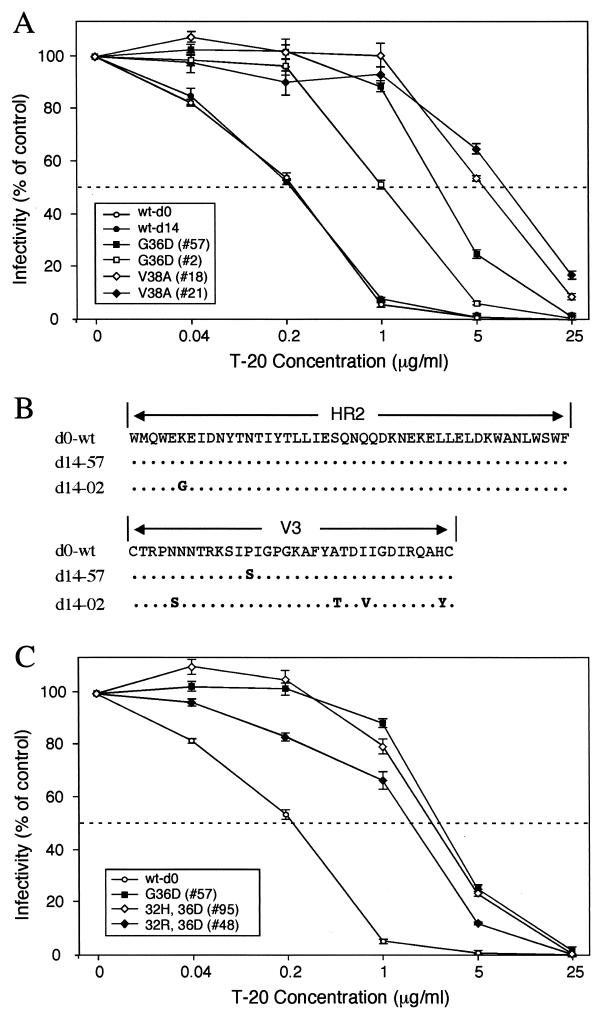
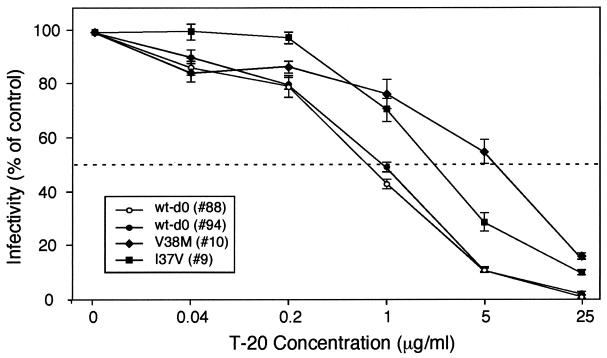
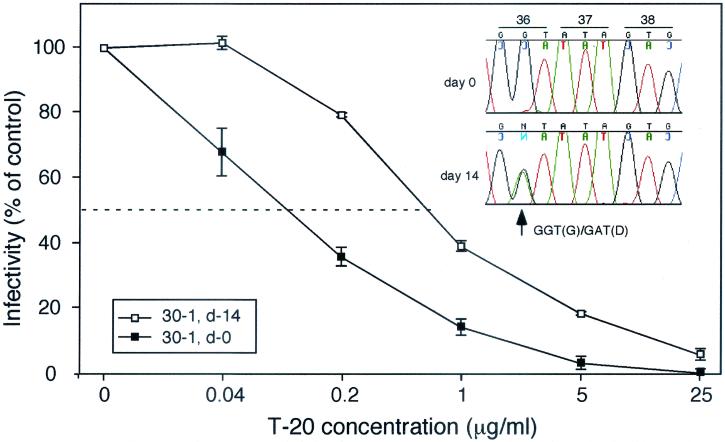
The synthetic peptide T-20 (enfuvirtide) represents the first of a new class of antiretroviral compounds to demonstrate in vivo potency by targeting a step in viral entry. T-20 inhibits a conformational change in the human immunodeficiency virus type 1 (HIV-1) transmembrane glycoprotein (gp41) that is required for fusion between HIV-1 and target cell membranes. The initial phase I clinical trial of T-20 treatment for HIV-infected patients thus provided a unique opportunity to evaluate the emergence of resistant virus in vivo to this novel class of antiretroviral agents. All four patients who received an intermediate dose of T-20 (30 mg twice daily) had an initial decline in plasma viral load over the first 10 days but a rising trend by day 14, suggestive of selection for resistant virus. Plasma virus derived from patients enrolled in all dosage groups of the phase I T-20 trial was analyzed by population sequencing before and after treatment. While no mutations were found within a highly conserved 3-amino-acid sequence (GIV) known to be critical for fusion at baseline, after 14 days of therapy, virus from one patient in the 30-mg dose group (30-1) developed a mutation in this motif, specifically an aspartic acid (D) substitution for glycine (G) at position 36. Multiple env clones were derived from the plasma virus of all four patients in the 30-mg dosage group. Sequence analysis of 49 clones derived from the plasma of patient 30-1 on day 14 revealed that 25 clones contained the G36D mutation, while 8 contained the V38A mutation. Dual mutations involving G36D and other residues within the HR1 domain were also identified. In 5 of the 49 env clones, other mutations involving residues 32 (Q32R or Q32H) and 39 (Q39R) were found in combination with G36D. Cloned env sequences derived from the plasma virus of subject 30-3 also had single mutations in the GIV sequence (V38M and I37V) detectable following therapy with T-20. The plasma virus from subjects 30-2 and 30-4 did not contain changes within the GIV sequence. To analyze the biological resistance properties of these mutations, we developed a novel single-cycle HIV-1 entry assay using JC53BL cells which express beta-galactosidase and luciferase under control of the HIV-1 long terminal repeat. Full-length env clones were derived from the plasma virus of patients 30-1 and 30-3 and used to generate pseudotyped virus stocks. The mean 50% inhibition concentrations (IC(50)s) for mutants G36D and V38A (patient 30-1) were 2.3 microg/ml and 11.2 microg/ml, respectively, statistically significant increases of 9.1- and 45-fold, respectively, compared with those of wild-type Env. The IC(50) for the V38 M mutation (patient 30-3) was 7.6 microg/ml, an 8-fold increase compared with that of the wild type. The I37V mutation resulted in an IC(50) 3.2-fold greater than that of the wild type. Envs with double mutations (Q32R plus G36D and Q32H plus G36D) exhibited a level of resistance similar to that of G36D alone. These findings provide the first evidence for the rapid emergence of clinical resistance to a novel class of HIV-1 entry inhibitors and may be relevant to future treatment strategies involving these agents.
Figures
References
-
- Chan, D. C., D. Fass, J. M. Berger, and P. S. Kim. 1997. Core structure of gp41 from the HIV envelope glycoprotein. Cell 89:263-273. - PubMed
-
- Condra, J. H., et al. 1995. In vivo emergence of HIV-1 variants resistant to multiple protease inhibitors. Nature 374:569-571. - PubMed
-
- Derdeyn, C. A., J. M. Decker, J. N. Sfakianos, Z. Zhang, W. A. O'Brien, L. Ratner, G. M. Shaw, and E. Hunter. 2001. Sensitivity of human immunodeficiency virus type 1 to fusion inhibitors targeted to the gp41 first heptad repeat involves distinct regions of gp41 and is consistently modulated by gp120 interactions with the coreceptor. J. Virol. 75:8605-8614. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
