

Modulation of human immunodeficiency virus 1 replication by interferon regulatory factors
- PMID: 12021315
- PMCID: PMC2193759
- DOI: 10.1084/jem.20010753
Modulation of human immunodeficiency virus 1 replication by interferon regulatory factors
Abstract
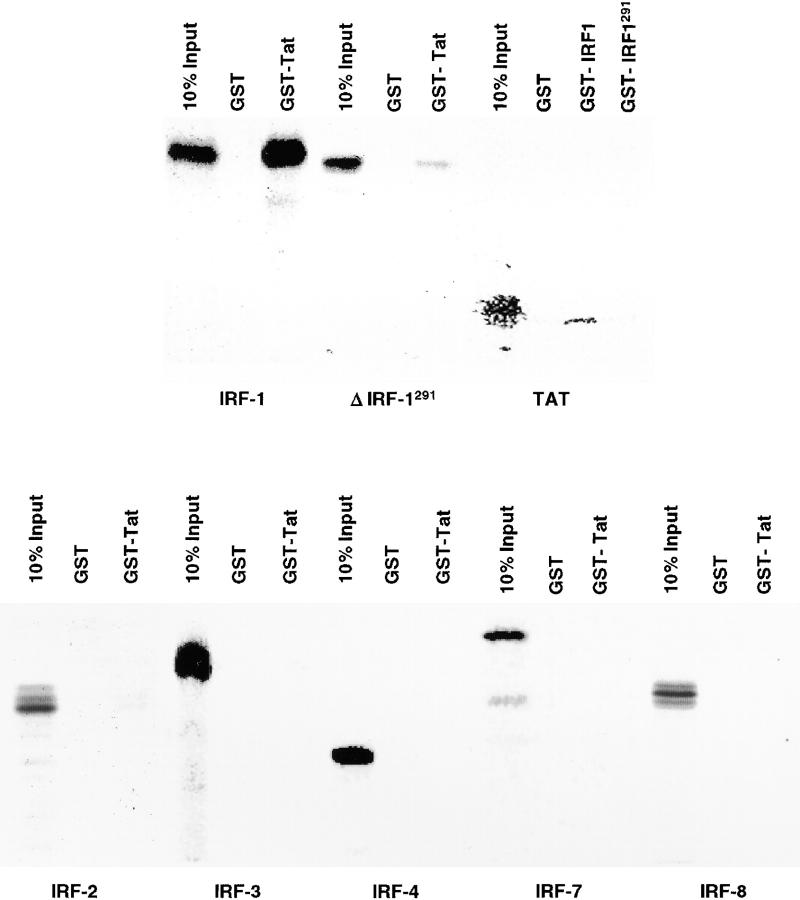
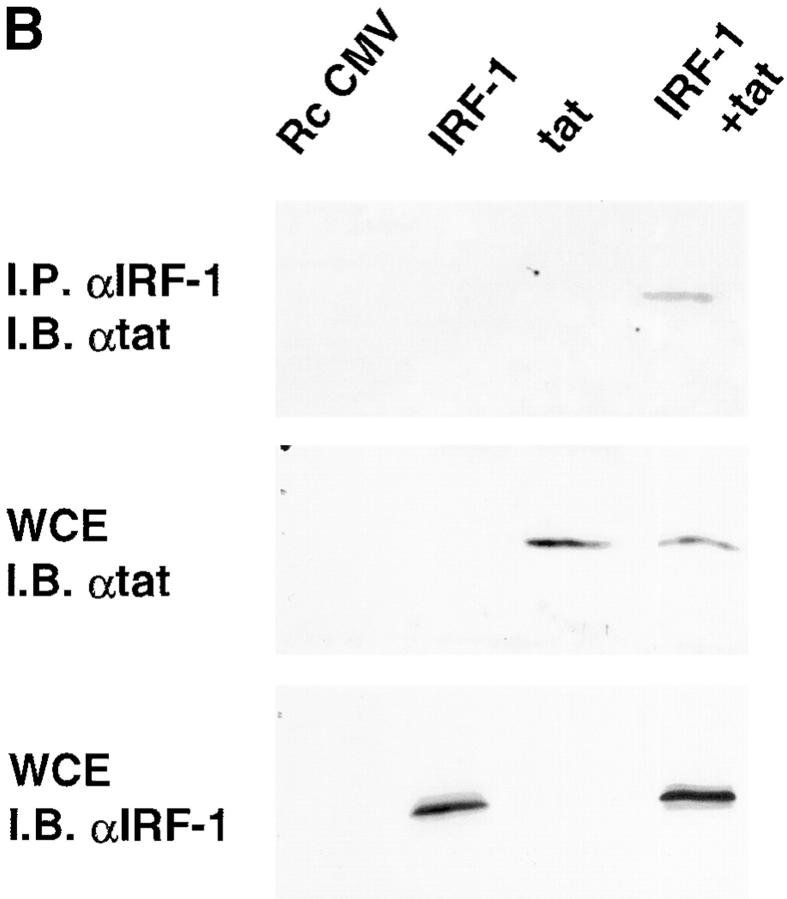
Transcription of the human immunodeficiency virus (HIV)-1 is controlled by the cooperation of virally encoded and host regulatory proteins. The Tat protein is essential for viral replication, however, expression of Tat after virus entry requires HIV-1 promoter activation. A sequence in the 5' HIV-1 LTR, containing a binding site for transcription factors of the interferon regulatory factors (IRF) family has been suggested to be critical for HIV-1 transcription and replication. Here we show that IRF-1 activates HIV-1 LTR transcription in a dose-dependent fashion and in the absence of Tat. This has biological significance since IRF-1 is produced early upon virus entry, both in cell lines and in primary CD4+ T cells, and before expression of Tat. IRF-1 also cooperates with Tat in amplifying virus gene transcription and replication. This cooperation depends upon a physical interaction that is blocked by overexpression of IRF-8, the natural repressor of IRF-1, and, in turn is released by overexpression of IRF-1. These data suggest a key role of IRF-1 in the early phase of viral replication and/or during viral reactivation from latency, when viral transactivators are absent or present at very low levels, and suggest that the interplay between IRF-1 and IRF-8 may play a key role in virus latency.
Figures


















Similar articles
-
A soluble factor(s) secreted from CD8(+) T lymphocytes inhibits human immunodeficiency virus type 1 replication through STAT1 activation.J Virol. 2002 Jan;76(2):569-81. doi: 10.1128/jvi.76.2.569-581.2002. J Virol. 2002. PMID: 11752148 Free PMC article.
-
Analysis of the HIV-1 LTR NF-kappaB-proximal Sp site III: evidence for cell type-specific gene regulation and viral replication.Virology. 2000 Sep 1;274(2):262-77. doi: 10.1006/viro.2000.0476. Virology. 2000. PMID: 10964770
-
Effect of SWI/SNF chromatin remodeling complex on HIV-1 Tat activated transcription.Retrovirology. 2006 Aug 7;3:48. doi: 10.1186/1742-4690-3-48. Retrovirology. 2006. PMID: 16893449 Free PMC article.
-
IRF regulation of HIV-1 long terminal repeat activity.J Interferon Cytokine Res. 2002 Jan;22(1):27-37. doi: 10.1089/107999002753452638. J Interferon Cytokine Res. 2002. PMID: 11846973 Review.
-
[Regulation of transcription in different HIV-1 subtypes].Enferm Infecc Microbiol Clin. 2005 Mar;23(3):156-62. doi: 10.1157/13072166. Enferm Infecc Microbiol Clin. 2005. PMID: 15757588 Review. Spanish.
Cited by
-
Genetic variability of the U5 and downstream sequence of major HIV-1 subtypes and circulating recombinant forms.Sci Rep. 2020 Aug 6;10(1):13214. doi: 10.1038/s41598-020-70083-1. Sci Rep. 2020. PMID: 32764600 Free PMC article.
-
Critical role of IRF-8 in negative regulation of TLR3 expression by Src homology 2 domain-containing protein tyrosine phosphatase-2 activity in human myeloid dendritic cells.J Immunol. 2011 Feb 15;186(4):1951-62. doi: 10.4049/jimmunol.1000918. Epub 2011 Jan 10. J Immunol. 2011. PMID: 21220691 Free PMC article.
-
Hydrogen sulfide blocks HIV rebound by maintaining mitochondrial bioenergetics and redox homeostasis.Elife. 2021 Nov 18;10:e68487. doi: 10.7554/eLife.68487. Elife. 2021. PMID: 34792020 Free PMC article.
-
A functional screen identifies transcriptional networks that regulate HIV-1 and HIV-2.Proc Natl Acad Sci U S A. 2021 Mar 16;118(11):e2012835118. doi: 10.1073/pnas.2012835118. Proc Natl Acad Sci U S A. 2021. PMID: 33836568 Free PMC article.
-
Junin Virus Triggers Macrophage Activation and Modulates Polarization According to Viral Strain Pathogenicity.Front Immunol. 2019 Oct 22;10:2499. doi: 10.3389/fimmu.2019.02499. eCollection 2019. Front Immunol. 2019. PMID: 31695702 Free PMC article.
References
-
- Cullen, B.R. 1991. Regulation of HIV-1 gene expression. FASEB J. 5:2361–2368. - PubMed
-
- Jones, K.A., and B.M. Peterlin. 1994. Control of RNA initiation and elongation at the HIV-1 promoter. Annu. Rev. Biochem. 63:717–743. - PubMed
-
- Gaynor, R. 1992. Cellular transcription factors involved in the regulation of HIV-1 gene expression. AIDS. 6:347–363. - PubMed
-
- el Kharroubi, A., and E. Verdin. 1994. Protein-DNA interactions within DNase I-hypersensitive sites located downstream of the HIV-1 promoter. J. Biol. Chem. 269:19916–19924. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
