

Protein-protein docking with multiple residue conformations and residue substitutions
- PMID: 12021438
- PMCID: PMC2373613
- DOI: 10.1110/ps.2830102
Protein-protein docking with multiple residue conformations and residue substitutions
Abstract
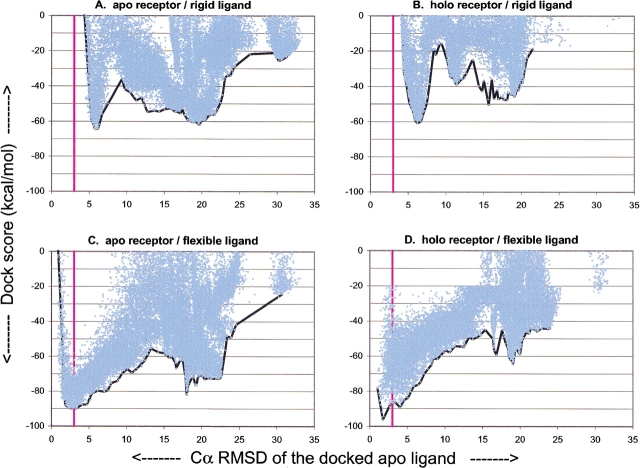
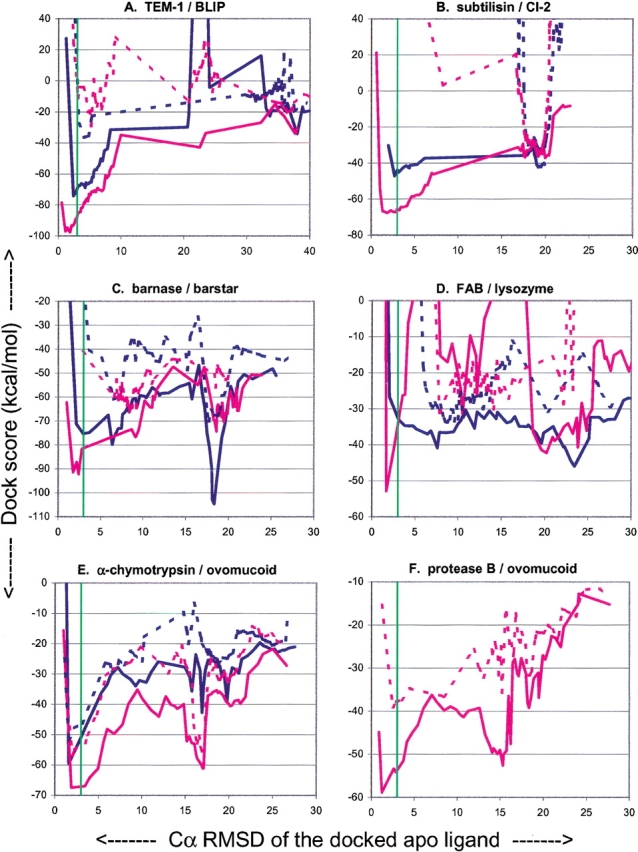
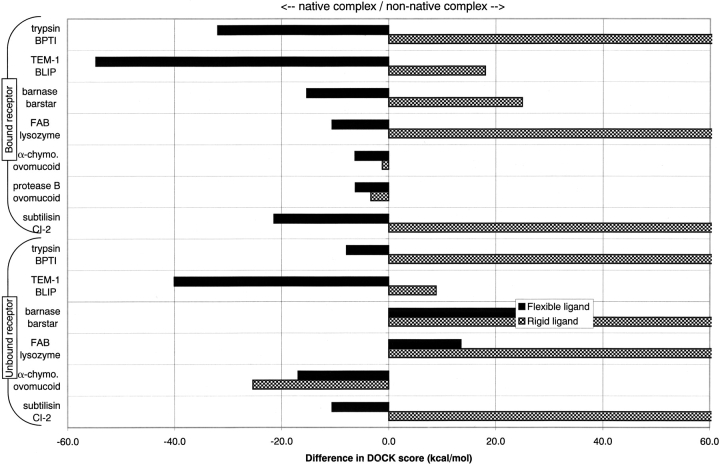
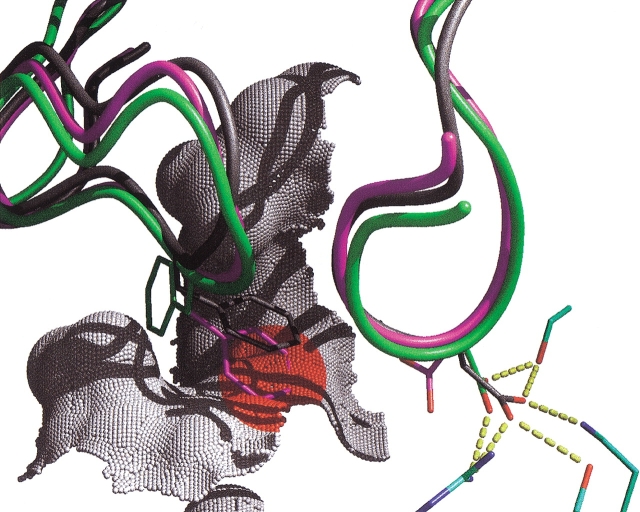
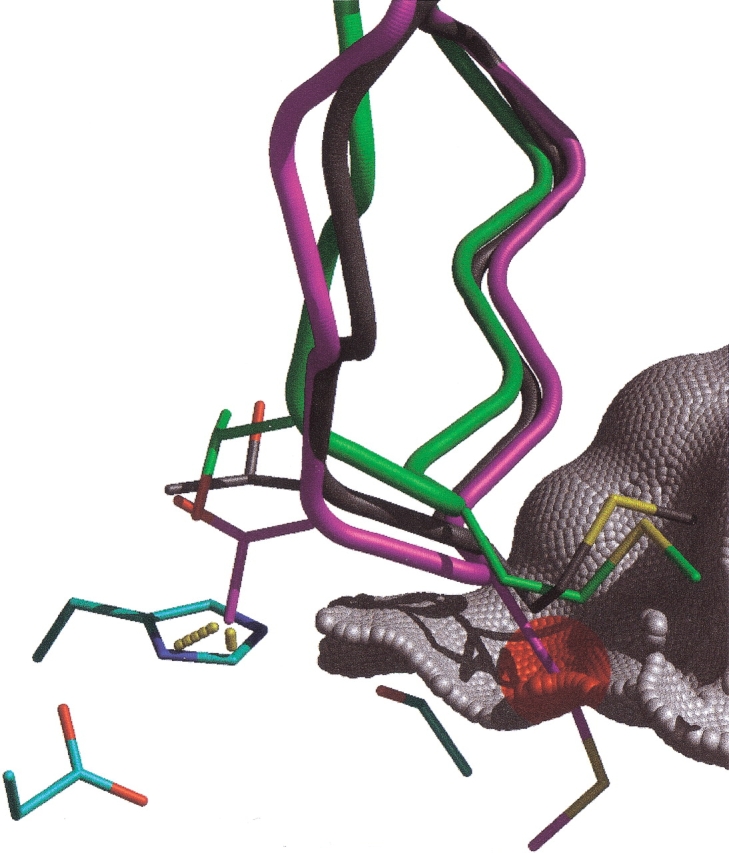
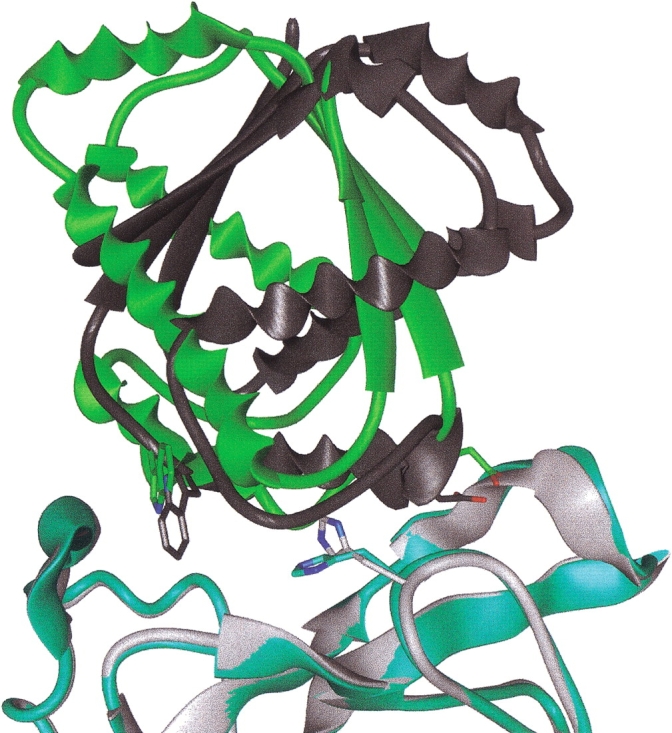
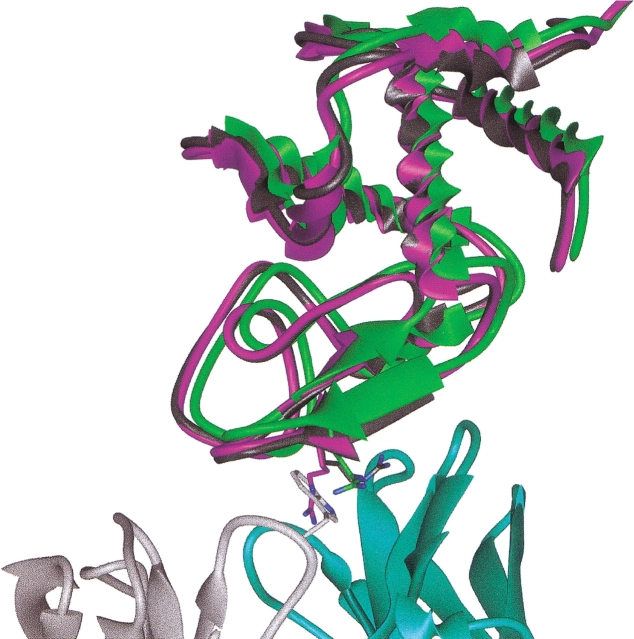
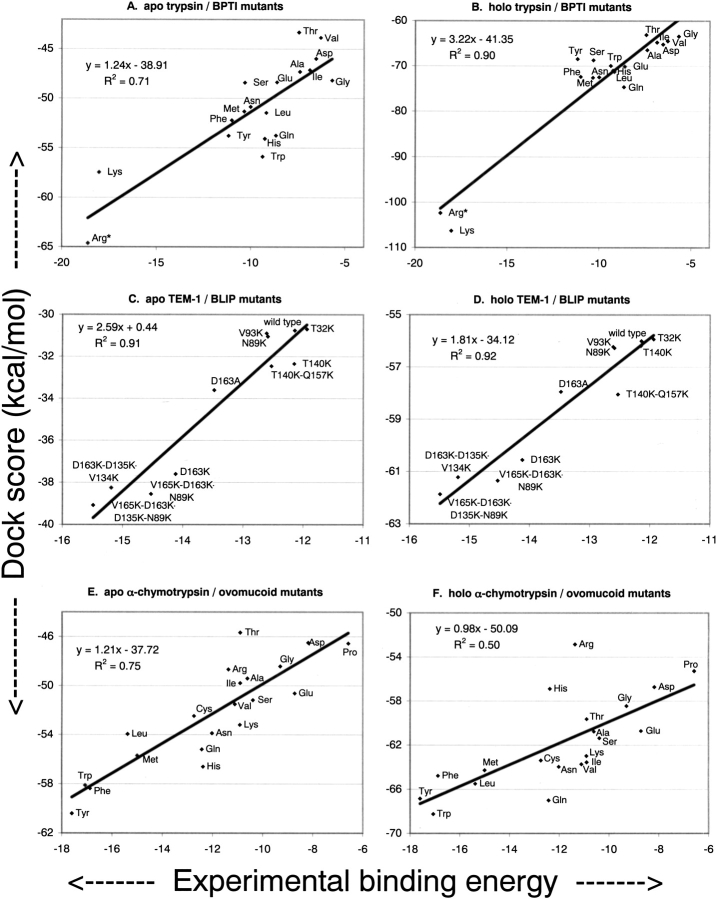
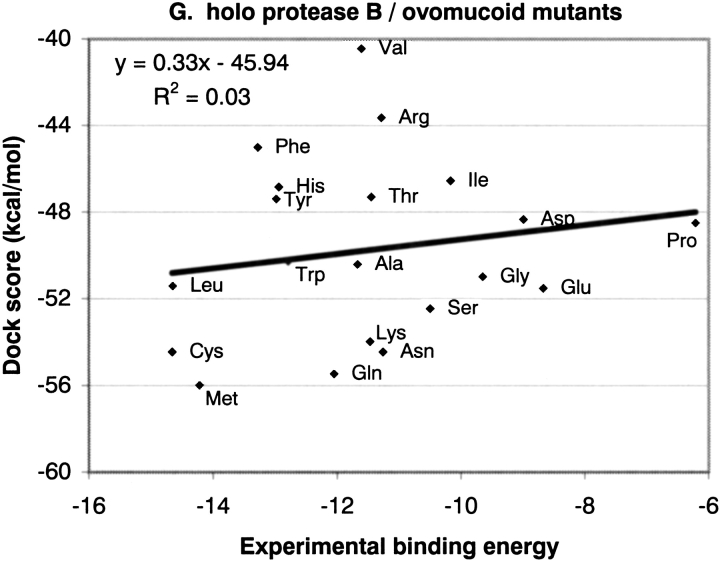
The protein docking problem has two major aspects: sampling conformations and orientations, and scoring them for fit. To investigate the extent to which the protein docking problem may be attributed to the sampling of ligand side-chain conformations, multiple conformations of multiple residues were calculated for the uncomplexed (unbound) structures of protein ligands. These ligand conformations were docked into both the complexed (bound) and unbound conformations of the cognate receptors, and their energies were evaluated using an atomistic potential function. The following questions were considered: (1) does the ensemble of precalculated ligand conformations contain a structure similar to the bound form of the ligand? (2) Can the large number of conformations that are calculated be efficiently docked into the receptors? (3) Can near-native complexes be distinguished from non-native complexes? Results from seven test systems suggest that the precalculated ensembles do include side-chain conformations similar to those adopted in the experimental complexes. By assuming additivity among the side chains, the ensemble can be docked in less than 12 h on a desktop computer. These multiconformer dockings produce near-native complexes and also non-native complexes. When docked against the bound conformations of the receptors, the near-native complexes of the unbound ligand were always distinguishable from the non-native complexes. When docked against the unbound conformations of the receptors, the near-native dockings could usually, but not always, be distinguished from the non-native complexes. In every case, docking the unbound ligands with flexible side chains led to better energies and a better distinction between near-native and non-native fits. An extension of this algorithm allowed for docking multiple residue substitutions (mutants) in addition to multiple conformations. The rankings of the docked mutant proteins correlated with experimental binding affinities. These results suggest that sampling multiple residue conformations and residue substitutions of the unbound ligand contributes to, but does not fully provide, a solution to the protein docking problem. Conformational sampling allows a classical atomistic scoring function to be used; such a function may contribute to better selectivity between near-native and non-native complexes. Allowing for receptor flexibility may further extend these results.
Figures
References
-
- Buckle, A.M., Schreiber, G., and Fersht, A.R. 1994. Protein–protein recognition: Crystal structural analysis of a barnase–barstar complex at 2.0-A resolution. Biochemistry 33 8878–8889. - PubMed
-
- Camacho, C.J., Gatchell, D.W., Kimura, S.R., and Vajda, S. 2000. Scoring docked conformations generated by rigid-body protein–protein docking. Proteins 40 525–537. - PubMed
-
- Cherfils, J., Duquerroy, S., and Janin, J. 1991. Protein–protein recognition analyzed by docking simulation. Proteins 11 271–280. - PubMed
-
- Claussen, H., Buning, C., Rarey, M., and Lengauer, T. 2001. FlexE: Efficient molecular docking considering protein structure variations. J. Mol. Biol. 308 377–395. - PubMed
-
- Connolly, M.L. 1986. Shape complementarity at the hemoglobin alpha 1 beta 1 subunit interface. Biopolymers 25 1229–1247. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
