

RIO: analyzing proteomes by automated phylogenomics using resampled inference of orthologs
- PMID: 12028595
- PMCID: PMC116988
- DOI: 10.1186/1471-2105-3-14
RIO: analyzing proteomes by automated phylogenomics using resampled inference of orthologs
Abstract
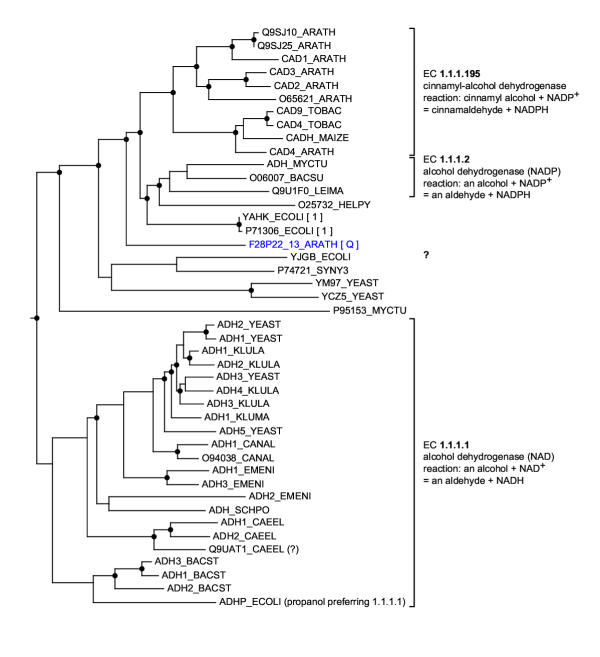
Background: When analyzing protein sequences using sequence similarity searches, orthologous sequences (that diverged by speciation) are more reliable predictors of a new protein's function than paralogous sequences (that diverged by gene duplication). The utility of phylogenetic information in high-throughput genome annotation ("phylogenomics") is widely recognized, but existing approaches are either manual or not explicitly based on phylogenetic trees.
Results: Here we present RIO (Resampled Inference of Orthologs), a procedure for automated phylogenomics using explicit phylogenetic inference. RIO analyses are performed over bootstrap resampled phylogenetic trees to estimate the reliability of orthology assignments. We also introduce supplementary concepts that are helpful for functional inference. RIO has been implemented as Perl pipeline connecting several C and Java programs. It is available at http://www.genetics.wustl.edu/eddy/forester/. A web server is at http://www.rio.wustl.edu/. RIO was tested on the Arabidopsis thaliana and Caenorhabditis elegans proteomes.
Conclusion: The RIO procedure is particularly useful for the automated detection of first representatives of novel protein subfamilies. We also describe how some orthologies can be misleading for functional inference.
Figures
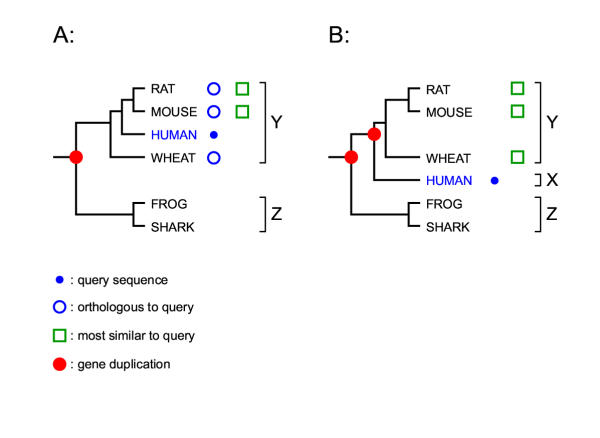
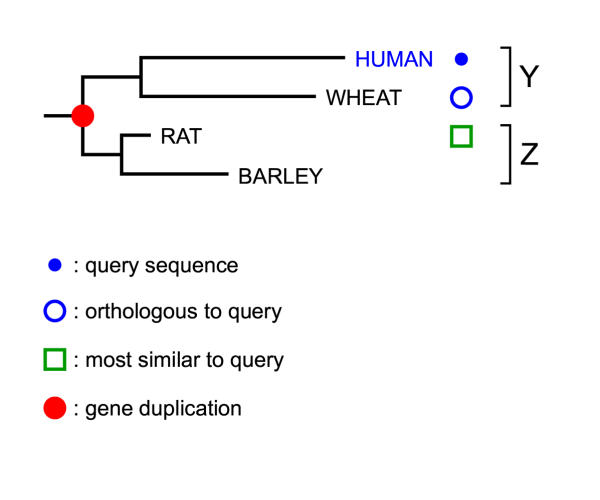
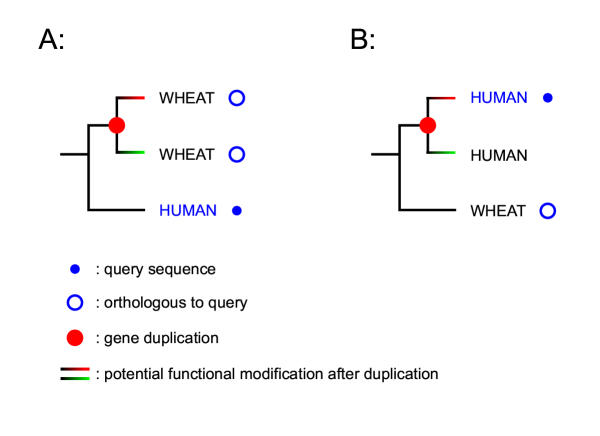
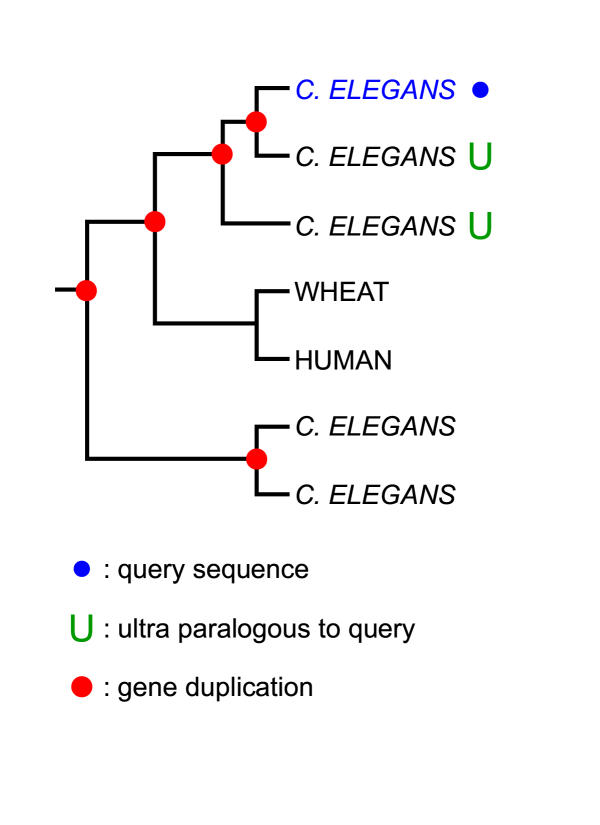


References
-
- Dayhoff MO. The origin and evolution of protein superfamilies. Fed Proc. 1976;35:2132–2138. - PubMed
-
- Ingram VM. Gene evolution and the haemoglobins. Nature. 1961;189:704–708. - PubMed
-
- Haldane JBS. The causes of evolution. New York and London: Harper & Brothers Publishers; 1932.
-
- Ohno S. Evolution by gene duplication. New York: Springer-Verlag; 1970.
-
- Galperin MY, Koonin EV. Sources of systematic error in functional annotation of genomes: domain rearrangement, non-orthologous gene displacement and operon disruption. In Silico Biol. 1998;1:55–67. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
