

Fast algorithms for large-scale genome alignment and comparison
- PMID: 12034836
- PMCID: PMC117189
- DOI: 10.1093/nar/30.11.2478
Fast algorithms for large-scale genome alignment and comparison
Abstract
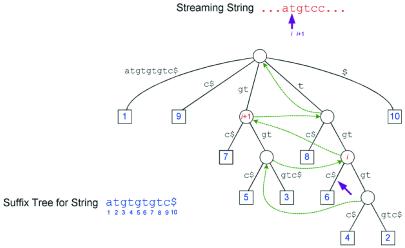
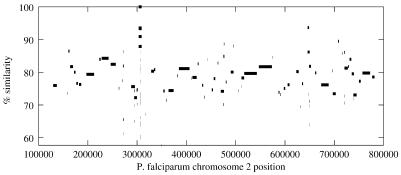
We describe a suffix-tree algorithm that can align the entire genome sequences of eukaryotic and prokaryotic organisms with minimal use of computer time and memory. The new system, MUMmer 2, runs three times faster while using one-third as much memory as the original MUMmer system. It has been used successfully to align the entire human and mouse genomes to each other, and to align numerous smaller eukaryotic and prokaryotic genomes. A new module permits the alignment of multiple DNA sequence fragments, which has proven valuable in the comparison of incomplete genome sequences. We also describe a method to align more distantly related genomes by detecting protein sequence homology. This extension to MUMmer aligns two genomes after translating the sequence in all six reading frames, extracts all matching protein sequences and then clusters together matches. This method has been applied to both incomplete and complete genome sequences in order to detect regions of conserved synteny, in which multiple proteins from one organism are found in the same order and orientation in another. The system code is being made freely available by the authors.
Figures
References
-
- Altschul S.F., Gish,W., Miller,W., Myers,E.W. and Lipman,D.J. (1990) Basic local alignment search tool. J. Mol. Biol., 215, 403–410. - PubMed
-
- Pearson W.R. (2000) Flexible sequence similarity searching with the FASTA3 program package. Methods Mol. Biol., 132, 185–219. - PubMed
-
- The Arabidopsis Genome Initiative (2000) Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature, 408,796–815. - PubMed
