

Comprehensive analysis of amino acid and nucleotide composition in eukaryotic genomes, comparing genes and pseudogenes
- PMID: 12034841
- PMCID: PMC117176
- DOI: 10.1093/nar/30.11.2515
Comprehensive analysis of amino acid and nucleotide composition in eukaryotic genomes, comparing genes and pseudogenes
Abstract
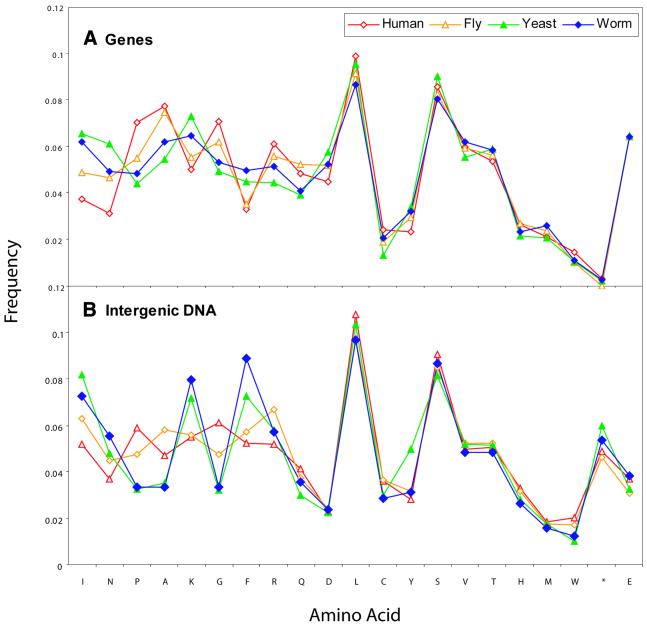
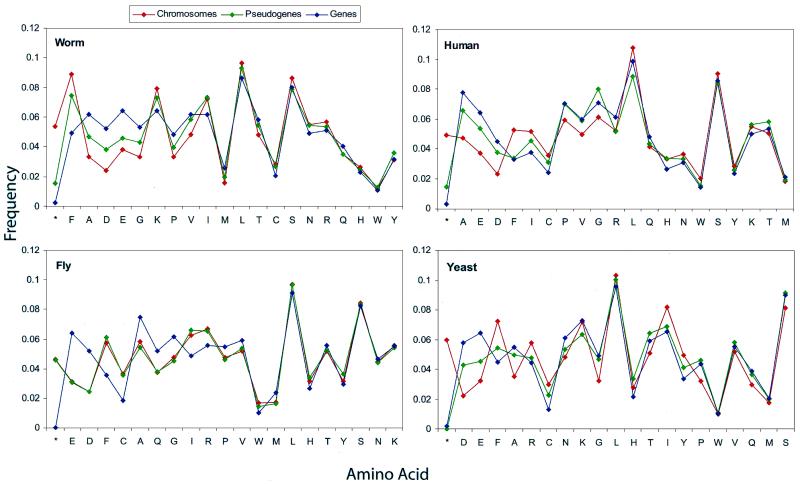
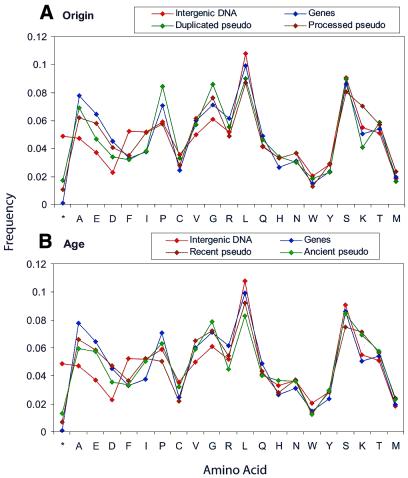
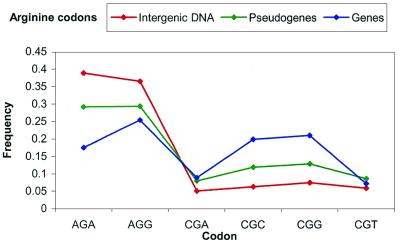
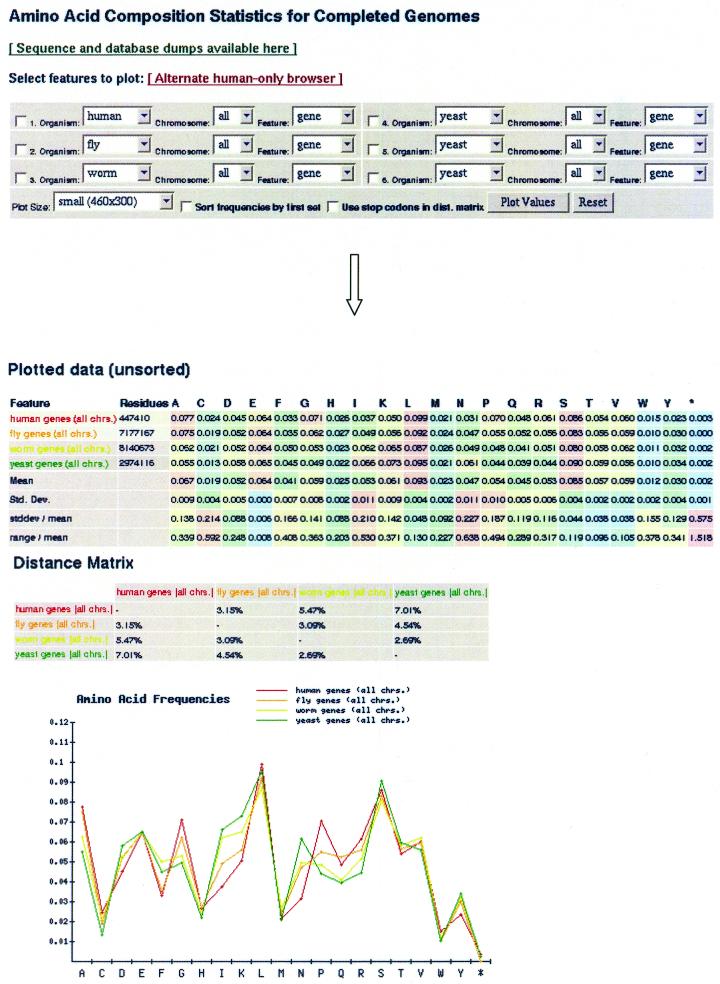
Based on searches for disabled homologs to known proteins, we have identified a large population of pseudogenes in four sequenced eukaryotic genomes-the worm, yeast, fly and human (chromosomes 21 and 22 only). Each of our nearly 2500 pseudogenes is characterized by one or more disablements mid-domain, such as premature stops and frameshifts. Here, we perform a comprehensive survey of the amino acid and nucleotide composition of these pseudogenes in comparison to that of functional genes and intergenic DNA. We show that pseudogenes invariably have an amino acid composition intermediate between genes and translated intergenic DNA. Although the degree of intermediacy varies among the four organisms, in all cases, it is most evident for amino acid types that differ most in occurrence between genes and intergenic regions. The same intermediacy also applies to codon frequencies, especially in the worm and human. Moreover, the intermediate composition of pseudogenes applies even though the composition of the genes in the four organisms is markedly different, showing a strong correlation with the overall A/T content of the genomic sequence. Pseudogenes can be divided into 'ancient' and 'modern' subsets, based on the level of sequence identity with their closest matching homolog (within the same genome). Modern pseudogenes usually have a much closer sequence composition to genes than ancient pseudogenes. Collectively, our results indicate that the composition of pseudogenes that are under no selective constraints progressively drifts from that of coding DNA towards non-coding DNA. Therefore, we propose that the degree to which pseudogenes approach a random sequence composition may be useful in dating different sets of pseudogenes, as well as to assess the rate at which intergenic DNA accumulates mutations. Our compositional analyses with the interactive viewer are available over the web at http://genecensus.org/pseudogene.
Figures
References
-
- Harrison P., Kumar,A., Lan,N., Echols,N., Snyder,M. and Gerstein,M. (2002) A small reservoir of disabled ORFs in the yeast genome and its implications for the dynamics of proteome evolution. J. Mol. Biol., 316, 409–419. - PubMed
-
- Harrison P.M. and Gerstein,M. (2002) Studying genomes through the aeons: protein families, pseudogenes and proteome evolution. J. Mol. Biol., in press. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
