

Pattern of organization of human mitochondrial pseudogenes in the nuclear genome
- PMID: 12045142
- PMCID: PMC1383742
- DOI: 10.1101/gr.227202
Pattern of organization of human mitochondrial pseudogenes in the nuclear genome
Abstract
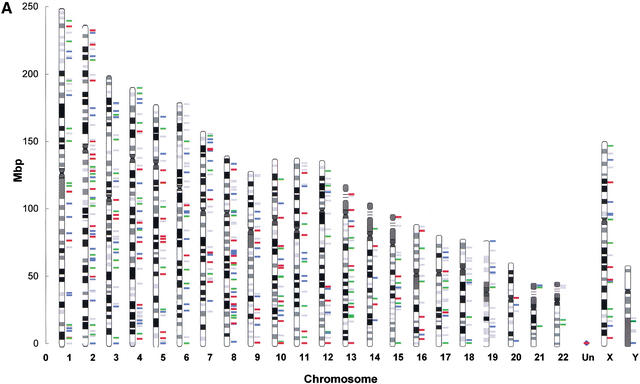
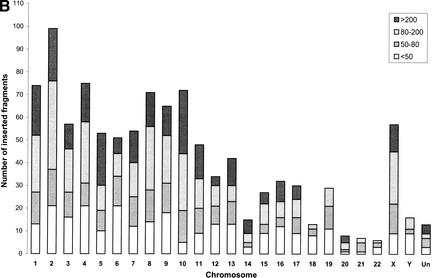
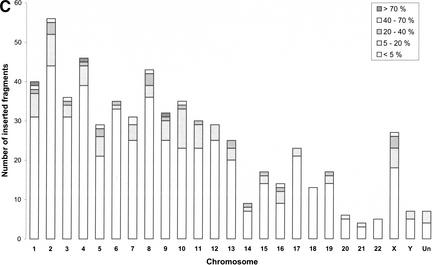
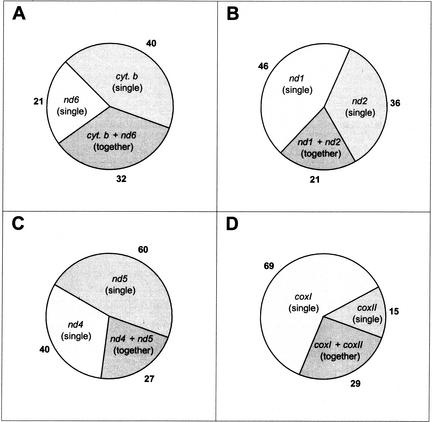
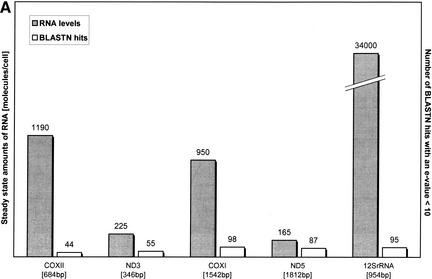
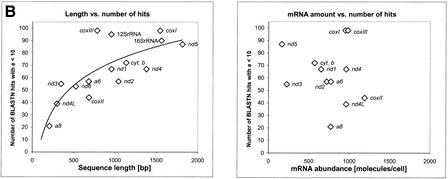
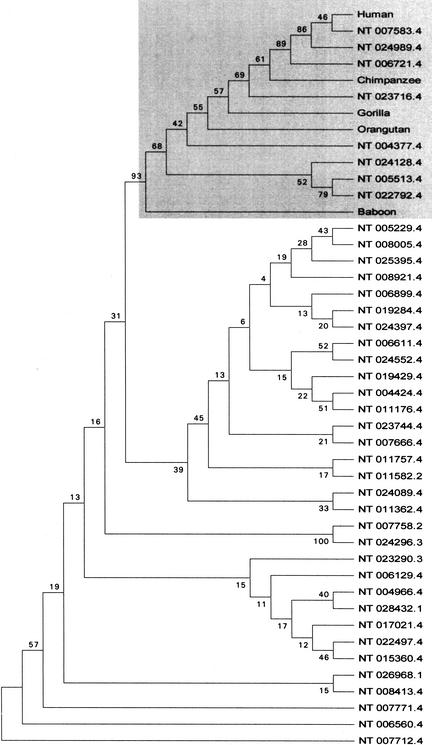
Mitochondrial pseudogenes in the human nuclear genome have been previously described, mostly as a source of artifacts during the analysis of the mitochondrial genome. With the availability of the complete human genome sequence, we performed a comprehensive analysis of mtDNA insertions into the nucleus. We found 612 independent integrations that are evenly distributed among all chromosomes as well as within each individual chromosome. The identified pseudogenes account for a content of at least 0.016% of the human nuclear DNA. Up to 30% of a chromosome's mtDNA pseudogene content is composed of fragments that encompass two or more adjacent mitochondrial genes, and we found no correlation between the abundance of mitochondrial transcripts and the multiplicity of integrations. These observations indicate that the migrations of mitochondrial DNA sequences to the nucleus were predominantly DNA mediated. Phylogenetic analysis of the mtDNA pseudogenes and mtDNA sequences of primates indicate a continuous transfer into the nucleus. Because of the limited window of opportunity for mtDNA transfer to the germline, sperm mtDNA, which is released from degenerating mitochondria after fertilization, could be an important source of nuclear mtDNA pseudogenes.
Figures

References
-
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. - PubMed
-
- Arctander P. Comparison of a mitochondrial gene and a corresponding nuclear pseudogene. Proc R Soc Lond B Biol Sci. 1995;262:13–19. - PubMed
-
- Attardi G, Chomyn A, King MP, Kruse B, Polosa PL, Murdter NN. Regulation of mitochondrial gene expression in mammalian cells. Biochem Soc Trans. 1990;18:509–513. - PubMed
-
- Bensasson D, Zhang D, Hartl DL, Hewitt GM. Mitochondrial pseudogenes: Evolution's misplaced witnesses. Trends Ecol Evol. 2001;16:314–321. - PubMed
-
- Blanchard JL, Lynch M. Organellar genes: Why do they end up in the nucleus? Trends Genet. 2000;16:315–320. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources