

ASK1 is essential for endoplasmic reticulum stress-induced neuronal cell death triggered by expanded polyglutamine repeats
- PMID: 12050113
- PMCID: PMC186318
- DOI: 10.1101/gad.992302
ASK1 is essential for endoplasmic reticulum stress-induced neuronal cell death triggered by expanded polyglutamine repeats
Abstract
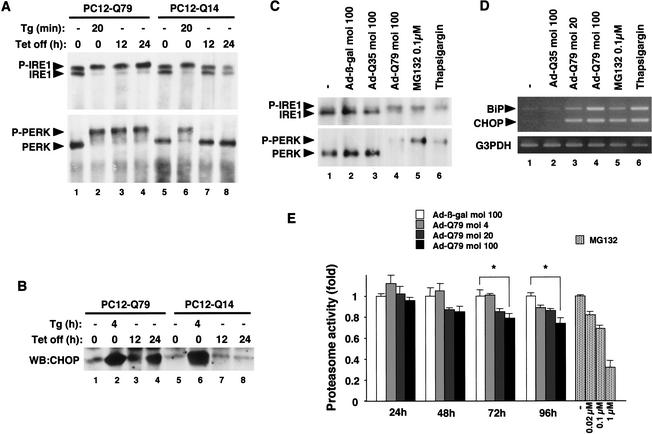
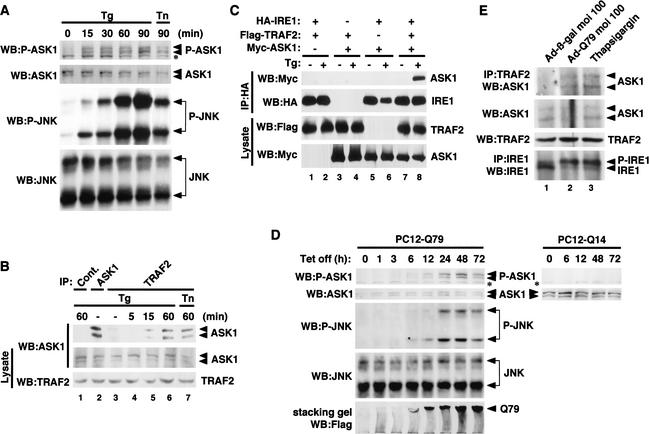
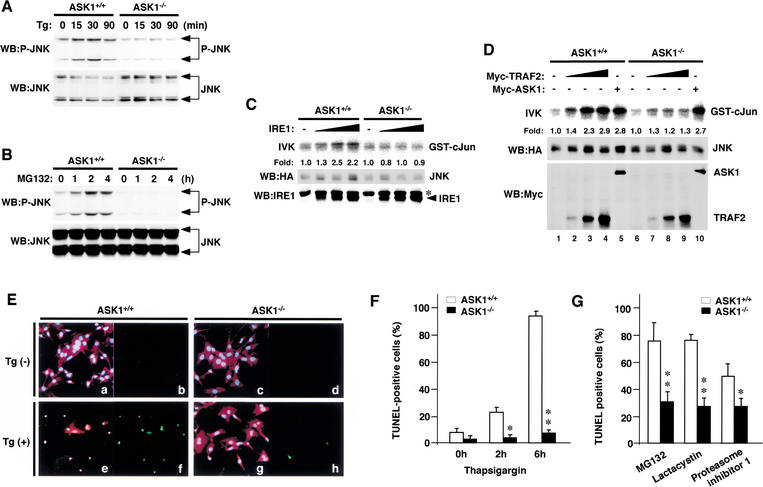
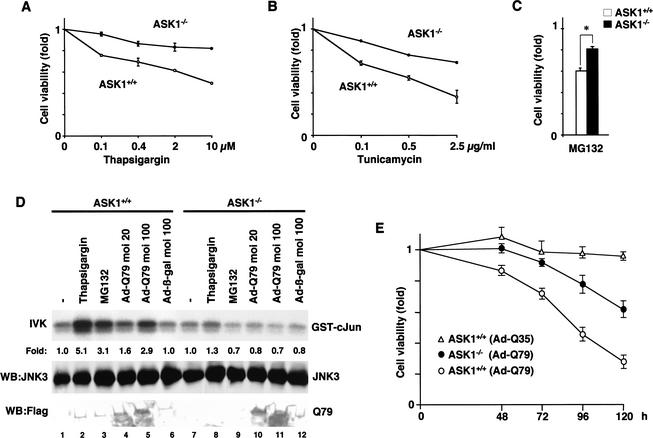
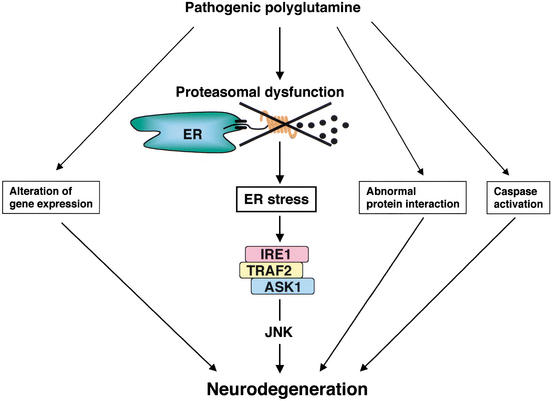
Expansion of CAG trinucleotide repeats that encode polyglutamine is the underlying cause of at least nine inherited human neurodegenerative disorders, including Huntington's disease and spinocerebellar ataxias. PolyQ fragments accumulate as aggregates in the cytoplasm and/or in the nucleus, and induce neuronal cell death. However, the molecular mechanism of polyQ-induced cell death is controversial. Here, we show the following: (1) polyQ with pathogenic repeat length triggers ER stress through proteasomal dysfunction; (2) ER stress activates ASK 1 through formation of an IRE1-TRAF2-ASK1 complex; and (3) ASK1(-/-) primary neurons are defective in polyQ-, proteasome inhibitor-, and ER stress-induced JNK activation and cell death. These findings suggest that ASK1 is a key element in ER stress-induced cell death that plays an important role in the neuropathological alterations in polyQ diseases.
Figures
References
-
- Bence NF, Sampat RM, Kopito RR. Impairment of the ubiquitin-proteasome system by protein aggregation. Science. 2001;292:1552–1555. - PubMed
-
- Bertolotti A, Zhang Y, Hendershot LM, Harding HP, Ron D. Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nat Cell Biol. 2000;2:326–332. - PubMed
-
- Bush KT, Goldberg AL, Nigam SK. Proteasome inhibition leads to a heat-shock response, induction of endoplasmic reticulum chaperones, and thermotolerance. J Biol Chem. 1997;272:9086–9092. - PubMed
-
- Delepine M, Nicolino M, Barrett T, Golamaully M, Lathrop GM, Julier C. EIF2AK3, encoding translation initiation factor 2-α kinase 3, is mutated in patients with Wolcott-Rallison syndrome. Nat Genet. 2000;25:406–409. - PubMed
-
- DiFiglia M, Sapp E, Chase KO, Davies SW, Bates GP, Vonsattel JP, Aronin N. Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science. 1997;277:1990–1993. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous