

Targeted inactivation of CTNNB1 reveals unexpected effects of beta-catenin mutation
- PMID: 12060769
- PMCID: PMC123056
- DOI: 10.1073/pnas.082240999
Targeted inactivation of CTNNB1 reveals unexpected effects of beta-catenin mutation
Abstract
Inactivating mutations of the adenomatous polyposis coli gene (APC) or activating mutations of the beta-catenin gene (CTNNB1) initiate colorectal neoplasia. To address the biochemical and physiologic effects of mutant beta-catenin, we disrupted either the mutant or wild-type CTNNB1 allele in a human colorectal cancer cell line. Cells with only wild-type beta-catenin had decreased colony-forming ability when plated at low density, although their growth was similar to that of parental cells when passaged under routine conditions. Immunohistochemistry and cell-fractionation studies suggested that mutant beta-catenin activity was distinguished primarily by cellular localization and not by protein degradation. Surprisingly, we found mutant beta-catenin bound less well to E-cadherin than did wild-type beta-catenin, and the membranous localization of wild-type and mutant beta-catenin was accordingly distinct. These findings pose several challenges to current models of APC/beta-catenin function.
Figures
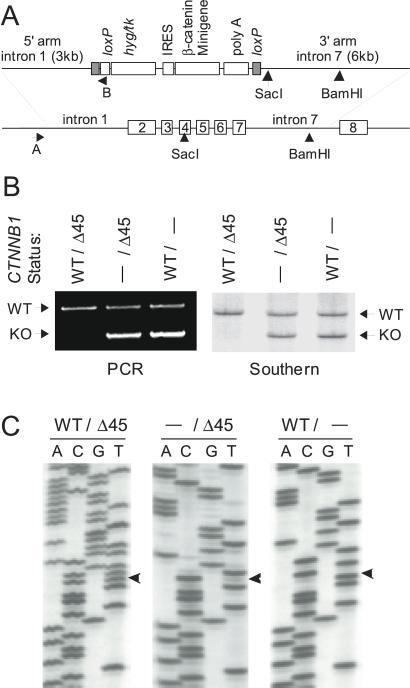
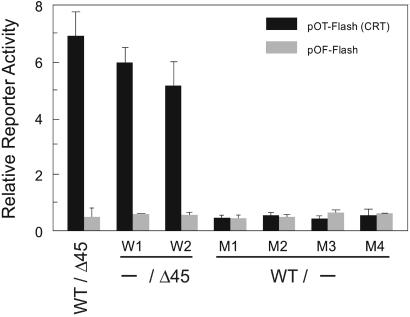
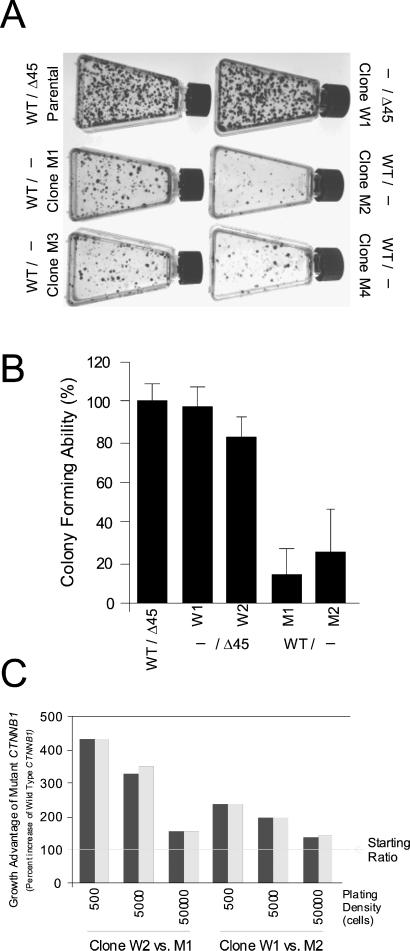
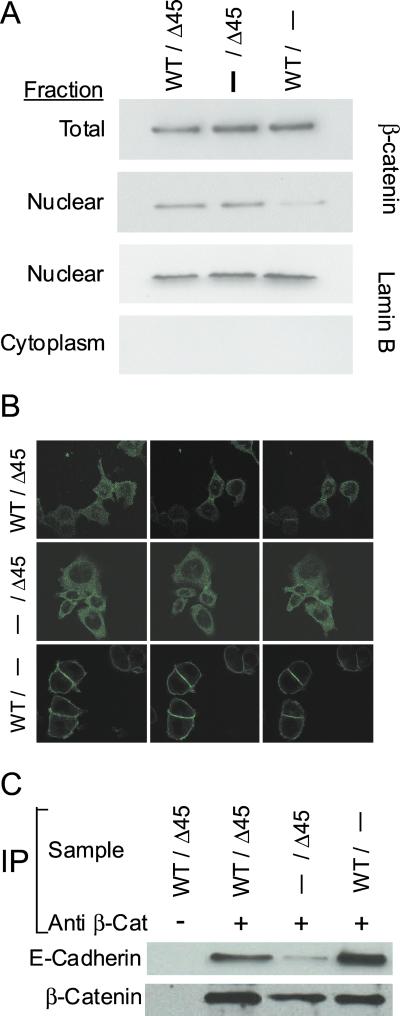
References
-
- American Cancer Society. Cancer Facts & Figures. Atlanta, GA: Am. Cancer Soc.; 2001.
-
- Kinzler K W, Vogelstein B. In: The Genetic Basis of Human Cancer. Vogelstein B, Kinzler K W, editors. New York: McGraw–Hill; 2002. pp. 565–587.
-
- Rubinfeld B, Souza B, Albert I, Muller O, Chamberlain S H, Masiarz F R, Munemitsu S, Polakis P. Science. 1993;262:1731–1734. - PubMed
-
- Su L K, Vogelstein B, Kinzler K W. Science. 1993;262:1734–1737. - PubMed
-
- Morin P J, Sparks A B, Korinek V, Barker N, Clevers H, Vogelstein B, Kinzler K W. Science. 1997;275:1787–1790. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
