

INK4a-deficient human diploid fibroblasts are resistant to RAS-induced senescence
- PMID: 12065407
- PMCID: PMC126048
- DOI: 10.1093/emboj/cdf289
INK4a-deficient human diploid fibroblasts are resistant to RAS-induced senescence
Abstract
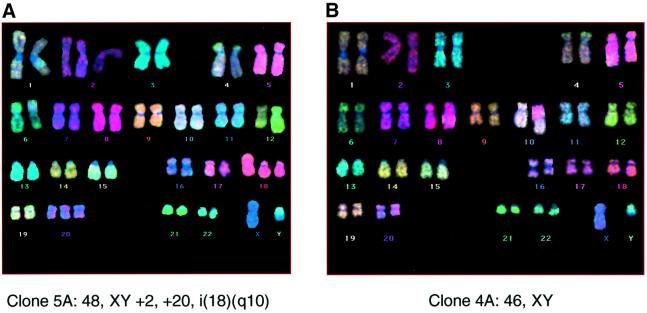
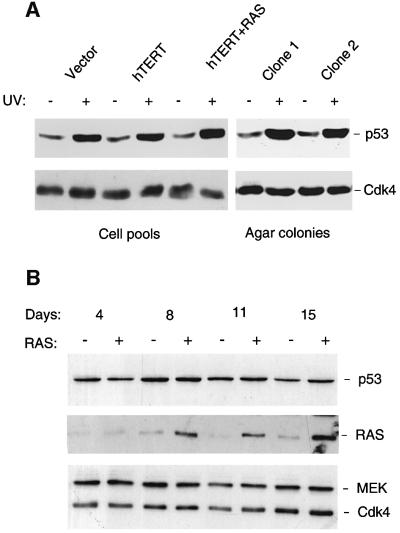
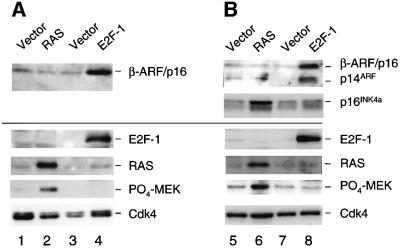
The CDKN2A tumour suppressor locus encodes two distinct proteins, p16(INK4a) and p14(ARF), both of which have been implicated in replicative senescence, the state of permanent growth arrest provoked in somatic cells by aberrant proliferative signals or by cumulative population doublings in culture. Here we describe primary fibroblasts from a member of a melanoma-prone family who is homozygous for an intragenic deletion in CDKN2A. Analyses of the resultant gene products imply that the cells are p16(INK4a) deficient but express physiologically relevant levels of a frameshift protein that retains the known functions of p14(ARF). Although they have a finite lifespan, the cells are resistant to arrest by oncogenic RAS. Indeed, ectopic expression of RAS and telomerase (hTERT) results in outgrowth of anchorage-independent colonies that have essentially diploid karyotypes and functional p53. We find that in human fibroblasts, ARF is not induced demonstrably by RAS, pointing to significant differences between the proliferative barriers implemented by the CDKN2A locus in different cell types or species.
Figures
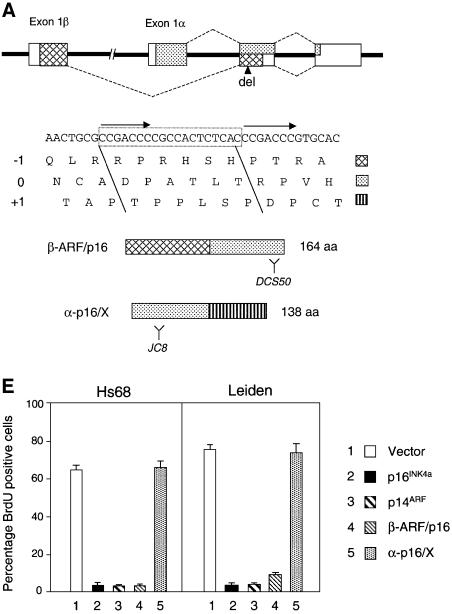
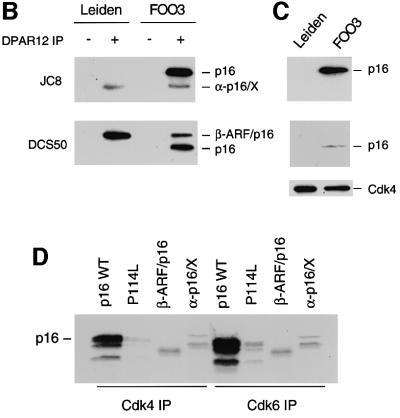
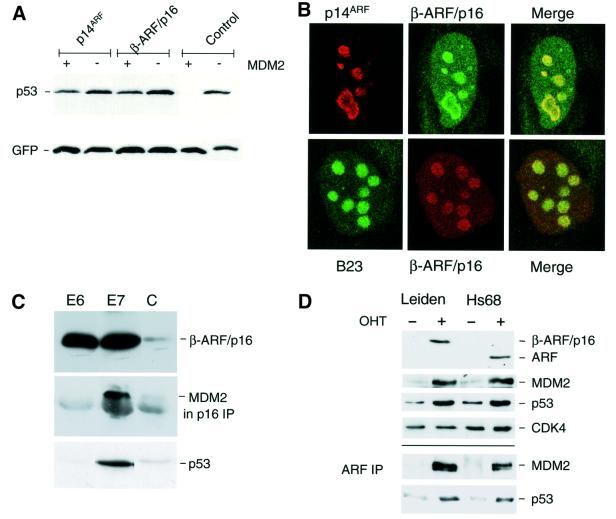
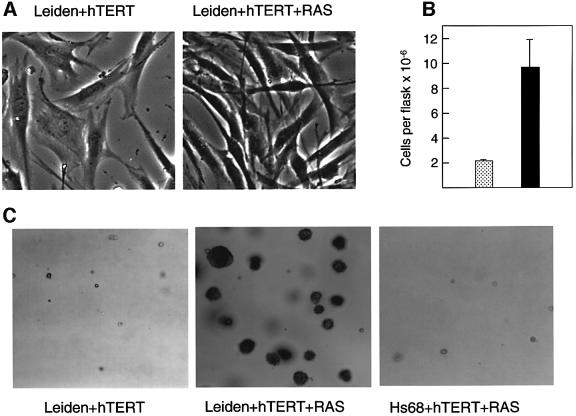
References
-
- Ashcroft M. and Vousden,K.H. (1999) Regulation of p53 stability. Oncogene, 18, 7637–7643. - PubMed
-
- Bodnar A.G. et al. (1998) Extension of life-span by introduction of telomerase into normal human cells. Science, 279, 349–352. - PubMed
-
- Brown J.P., Wei,W. and Sedivy,J.M. (1997) Bypass of senescence after disruption of p21CIP1/WAF1 gene in normal diploid human fibroblasts. Science, 277, 831–834. - PubMed
-
- Bunz F., Dutriaux,A., Lengauer,C., Waldman,T., Zhou,S., Brown,J.P., Sedivy,J.M., Kinzler,K.W. and Vogelstein,B. (1998) Requirement for p53 and p21 to sustain G2 arrest after DNA damage. Science, 282, 1497–1501. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
