

A novel uracil-DNA glycosylase with broad substrate specificity and an unusual active site
- PMID: 12065430
- PMCID: PMC126064
- DOI: 10.1093/emboj/cdf309
A novel uracil-DNA glycosylase with broad substrate specificity and an unusual active site
Abstract
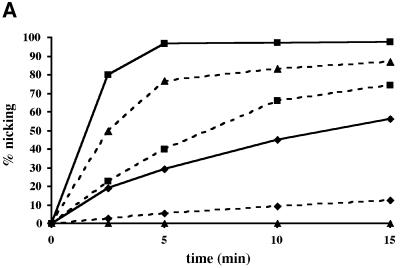
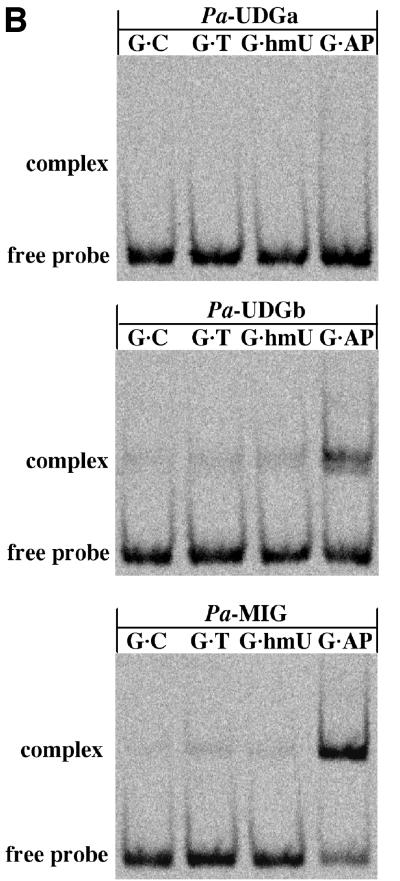
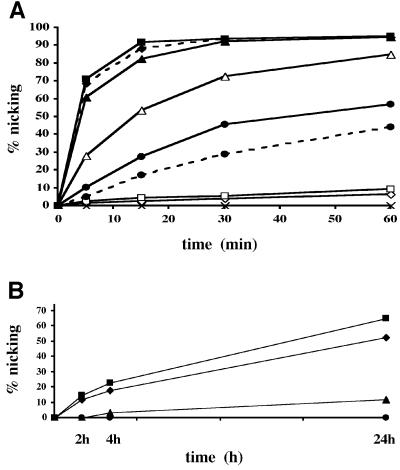
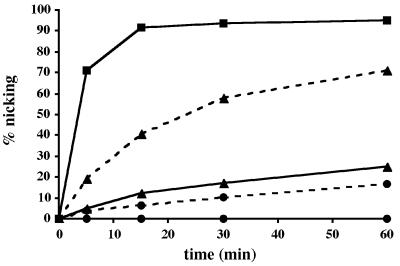
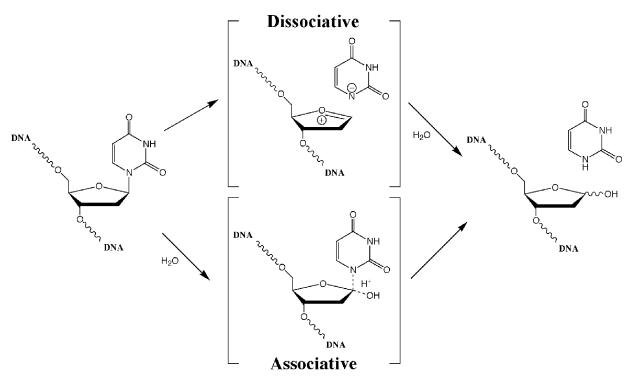
Uracil-DNA glycosylases (UDGs) catalyse the removal of uracil by flipping it out of the double helix into their binding pockets, where the glycosidic bond is hydrolysed by a water molecule activated by a polar amino acid. Interestingly, the four known UDG families differ in their active site make-up. The activating residues in UNG and SMUG enzymes are aspartates, thermostable UDGs resemble UNG-type enzymes, but carry glutamate rather than aspartate residues in their active sites, and the less active MUG/TDG enzymes contain an active site asparagine. We now describe the first member of a fifth UDG family, Pa-UDGb from the hyperthermophilic crenarchaeon Pyrobaculum aerophilum, the active site of which lacks the polar residue that was hitherto thought to be essential for catalysis. Moreover, Pa-UDGb is the first member of the UDG family that efficiently catalyses the removal of an aberrant purine, hypoxanthine, from DNA. We postulate that this enzyme has evolved to counteract the mutagenic threat of cytosine and adenine deamination, which becomes particularly acute in organisms living at elevated temperatures.
Figures



Similar articles
-
Characterization of a thermostable DNA glycosylase specific for U/G and T/G mismatches from the hyperthermophilic archaeon Pyrobaculum aerophilum.J Bacteriol. 2000 Mar;182(5):1272-9. doi: 10.1128/JB.182.5.1272-1279.2000. J Bacteriol. 2000. PMID: 10671447 Free PMC article.
-
Enzymology of base excision repair in the hyperthermophilic archaeon Pyrobaculum aerophilum.J Biol Chem. 2003 Jul 4;278(27):24563-76. doi: 10.1074/jbc.M302397200. Epub 2003 Apr 30. J Biol Chem. 2003. PMID: 12730226
-
Protein mimicry of DNA from crystal structures of the uracil-DNA glycosylase inhibitor protein and its complex with Escherichia coli uracil-DNA glycosylase.J Mol Biol. 1999 Mar 26;287(2):331-46. doi: 10.1006/jmbi.1999.2605. J Mol Biol. 1999. PMID: 10080896
-
Structure and function in the uracil-DNA glycosylase superfamily.Mutat Res. 2000 Aug 30;460(3-4):165-81. doi: 10.1016/s0921-8777(00)00025-2. Mutat Res. 2000. PMID: 10946227 Review.
-
Uracil in DNA--occurrence, consequences and repair.Oncogene. 2002 Dec 16;21(58):8935-48. doi: 10.1038/sj.onc.1205996. Oncogene. 2002. PMID: 12483510 Review.
Cited by
-
Processing of DNA lesions by archaeal DNA polymerases from Sulfolobus solfataricus.Nucleic Acids Res. 2003 Jul 15;31(14):4024-30. doi: 10.1093/nar/gkg447. Nucleic Acids Res. 2003. PMID: 12853619 Free PMC article.
-
Cytosine deamination is a major cause of baseline noise in next-generation sequencing.Mol Diagn Ther. 2014 Oct;18(5):587-93. doi: 10.1007/s40291-014-0115-2. Mol Diagn Ther. 2014. PMID: 25091469 Free PMC article.
-
Uracil-DNA glycosylase of Thermoplasma acidophilum directs long-patch base excision repair, which is promoted by deoxynucleoside triphosphates and ATP/ADP, into short-patch repair.J Bacteriol. 2011 Sep;193(17):4495-508. doi: 10.1128/JB.00233-11. Epub 2011 Jun 10. J Bacteriol. 2011. PMID: 21665970 Free PMC article.
-
Archaeal genome guardians give insights into eukaryotic DNA replication and damage response proteins.Archaea. 2014 Feb 20;2014:206735. doi: 10.1155/2014/206735. eCollection 2014. Archaea. 2014. PMID: 24701133 Free PMC article. Review.
-
Sulfolobus acidocaldarius UDG Can Remove dU from the RNA Backbone: Insight into the Specific Recognition of Uracil Linked with Deoxyribose.Genes (Basel). 2017 Jan 18;8(1):38. doi: 10.3390/genes8010038. Genes (Basel). 2017. PMID: 28106786 Free PMC article.
References
-
- Barrett T.E., Savva,R., Panayotou,G., Barlow,T., Brown,T., Jiricny,J. and Pearl,L.H. (1998) Crystal structure of a G:T/U mismatch-specific DNA glycosylase: mismatch recognition by complementary-strand interactions. Cell, 92, 117–129. - PubMed
-
- Brown T.C. and Brown-Luedi,M.L. (1989) G/U lesions are efficiently corrected to G/C in SV40 DNA. Mutat. Res., 227, 233–236. - PubMed
-
- Brown T.C. and Jiricny,J. (1987) A specific mismatch repair event protects mammalian cells from loss of 5-methylcytosine. Cell, 50, 945–950. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
