

Cocaine binds to a common site on open and inactivated human heart (Na(v)1.5) sodium channels
- PMID: 12068034
- PMCID: PMC2290378
- DOI: 10.1113/jphysiol.2001.016139
Cocaine binds to a common site on open and inactivated human heart (Na(v)1.5) sodium channels
Abstract
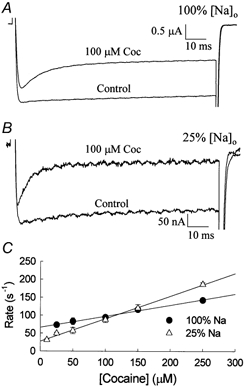
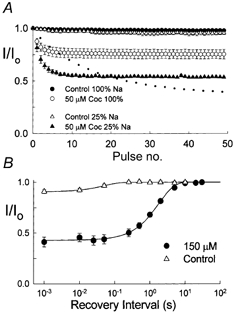
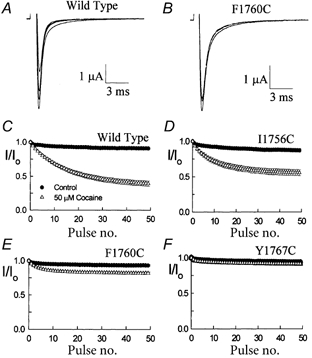
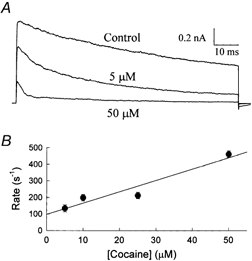
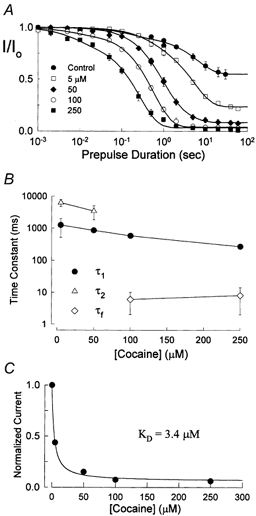
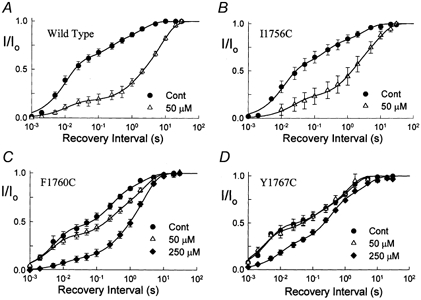
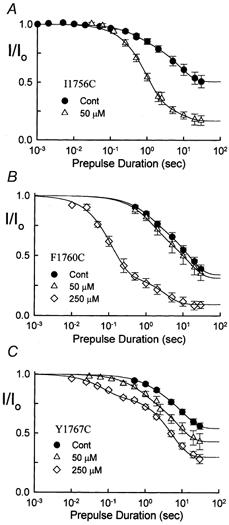
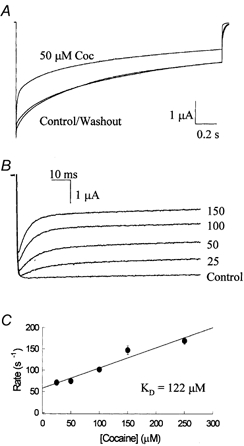
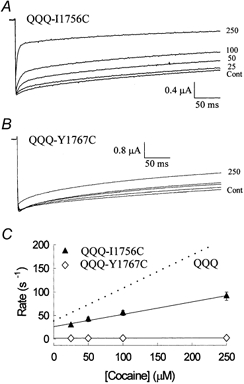
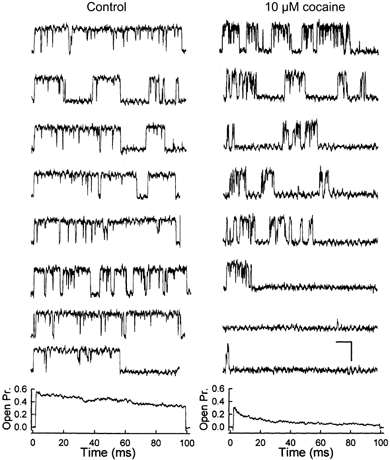
The inhibition by cocaine of the human heart Na+ channel (Na(v)1.5) heterologously expressed in Xenopus oocytes was investigated. Cocaine produced little tonic block of the resting channels but induced a characteristic, use-dependent inhibition during rapid, repetitive stimulation, suggesting that the drug preferentially binds to the open or inactivated states of the channel. To investigate further the state dependence, depolarizing pulses were used to inactivate the channels and promote cocaine binding. Cocaine produced a slow, concentration-dependent inhibition of inactivated channels, which had an apparent K(D) of 3.4 microM. Mutations of the interdomain III-IV linker that remove fast inactivation selectively abolished this high-affinity component of cocaine inhibition, which appeared to be linked to the fast inactivation of the channels. A rapid component of cocaine inhibition persisted in the inactivation-deficient mutant that was enhanced by depolarization and was sensitive to changes in the concentration of external Na+, properties that are consistent with a pore-blocking mechanism. Cocaine induced a use-dependent inhibition of the non-inactivating mutant and delayed the repriming at hyperpolarized voltages, indicating that the drug slowly dissociated when the channels were closed. Mutation of a conserved aromatic residue (Y1767) of the D4S6 segment weakened both the inactivation-dependent and the pore-blocking components of the cocaine inhibition. The data indicate that cocaine binds to a common site located within the internal vestibule and inhibits cardiac Na+ channels by blocking the pore and by stabilizing the channels in an inactivated state.
Figures
References
-
- Alpert LA, Fozzard HA, Hanck DA, Makielski JC. Is there a second external lidocaine binding site on mammalian cardiac cells? American Journal of Physiology. 1989;257:H79–84. - PubMed
-
- Bennett PB, Valenzuela C, Chen LQ, Kallen RG. On the molecular nature of the lidocaine receptor of cardiac Na+ channels. Modification of block by alterations in the alpha-subunit III-IV interdomain. Circulation Research. 1995;77:584–592. - PubMed
-
- Billman GE. Cocaine: a review of its toxic actions on cardiac function. Critical Review of Toxicology. 1995;25:113–132. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous