

Inducing tumor immunity through the selective engagement of activating Fcgamma receptors on dendritic cells
- PMID: 12070293
- PMCID: PMC2193555
- DOI: 10.1084/jem.20020338
Inducing tumor immunity through the selective engagement of activating Fcgamma receptors on dendritic cells
Abstract
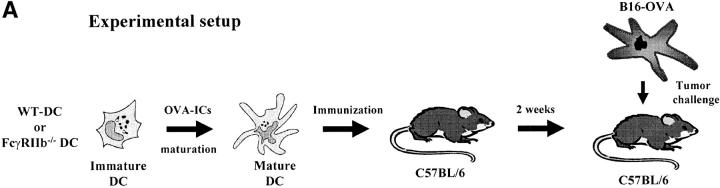
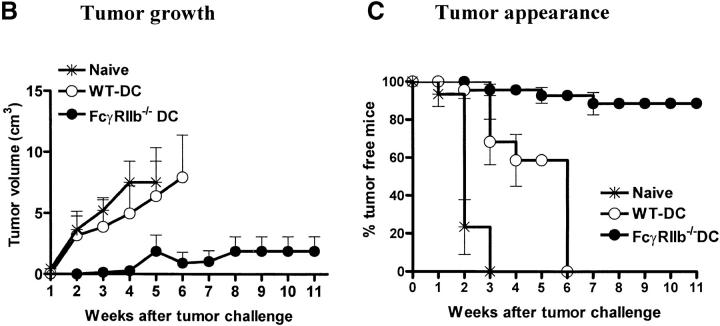
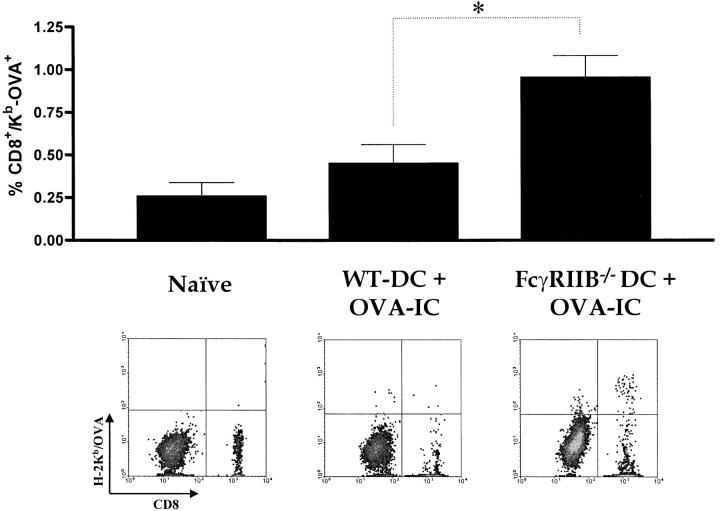
Induction of tumor-specific immunity requires that dendritic cells (DCs) efficiently capture and present tumor antigens to result in the expansion and activation of tumor-specific cytotoxic T cells. The transition from antigen capture to T cell stimulation requires a maturation signal; in its absence tolerance, rather than immunity may develop. While immune complexes (ICs) are able to enhance antigen capture, they can be poor at inducing DC maturation, naive T cell activation and protective immunity. We now demonstrate that interfering with the inhibitory signal delivered by FcgammaRIIB on DCs converts ICs to potent maturation agents and results in T cell activation. Applying this approach to immunization with DCs pulsed ex-vivo with ICs, we have generated antigen-specific CD8+ T cells in vivo and achieved efficient protective immunity in a murine melanoma model. These data imply that ICs may normally function to maintain tolerance through the binding to inhibitory FcgammaRs on DCs, but they can be converted to potent immunogenic stimuli by selective engagement of activating FcgammaRs. This mechanism suggests a novel approach to the development of tumor vaccines.
Figures
References
-
- LaTemple, D.C., J.T. Abrams, S.Y. Zhang, and U. Galili. 1999. Increased immunogenicity of tumor vaccines complexed with anti-Gal: studies in knockout mice for α1,3galactosyltransferase. Cancer Res. 59:3417–3423. - PubMed
-
- Banchereau, J., F. Briere, C. Caux, J. Davoust, S. Lebecque, Y.J. Liu, B. Pulendran, and K. Palucka. 2000. Immunobiology of dendritic cells. Annu. Rev. Immunol. 18:767–811. - PubMed
-
- Fong, L., and E.G. Engleman. 2000. Dendritic cells in cancer immunotherapy. Annu. Rev. Immunol. 18:245–273. - PubMed
-
- Lanzavecchia, A., and F. Sallusto. 2001. Regulation of T cell immunity by dendritic cells. Cell. 106:263–266. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
