

Autosomal dominant stapes ankylosis with broad thumbs and toes, hyperopia, and skeletal anomalies is caused by heterozygous nonsense and frameshift mutations in NOG, the gene encoding noggin
- PMID: 12089654
- PMCID: PMC379196
- DOI: 10.1086/342067
Autosomal dominant stapes ankylosis with broad thumbs and toes, hyperopia, and skeletal anomalies is caused by heterozygous nonsense and frameshift mutations in NOG, the gene encoding noggin
Abstract
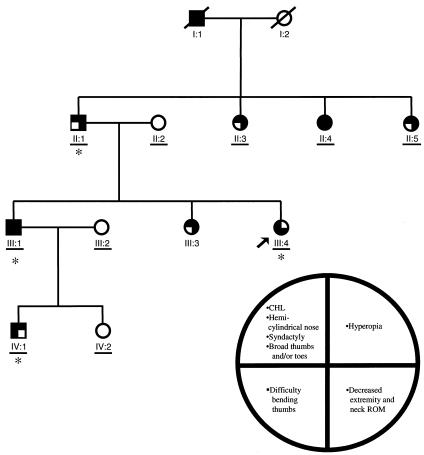
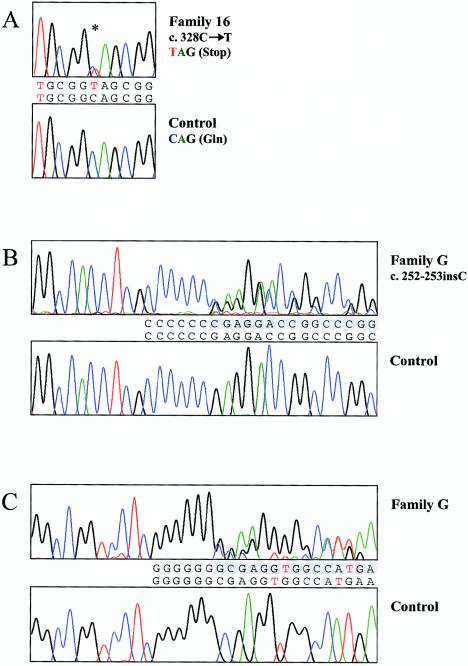
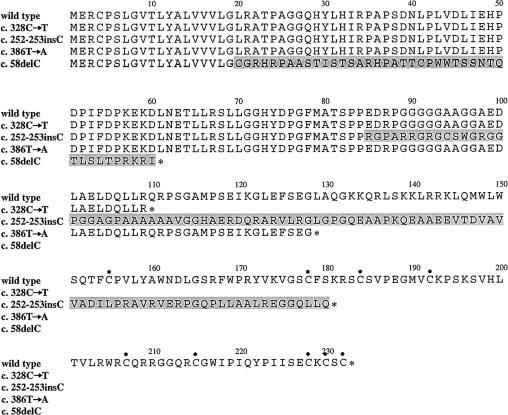
Although fixation of the stapes is usually progressive and secondary to otosclerosis, it may present congenitally, with other skeletal manifestations, as an autosomal dominant syndrome-such as proximal symphalangism (SYM1) or multiple-synostoses syndrome (SYNS1), both of which are caused by mutations in NOG, the gene encoding noggin. We describe a family that was ascertained to have nonsyndromic otosclerosis but was subsequently found to have a congenital stapes ankylosis syndrome that included hyperopia, a hemicylindrical nose, broad thumbs and great toes, and other minor skeletal anomalies but lacked symphalangism. A heterozygous nonsense NOG mutation-c.328C-->T (Q110X), predicted to truncate the latter half of the protein-was identified, and a heterozygous insertion in NOG-c.252-253insC, in which the frameshift is predicted to result in 96 novel amino acids before premature truncation-was identified in a previously described second family with a similar phenotype. In contrast to most NOG mutations that have been reported in kindreds with SYM1 and SYNS1, the mutations observed in these families with stapes ankylosis without symphalangism are predicted to disrupt the cysteine-rich C-terminal domain. These clinical and molecular findings suggest that (1) a broader range of conductive hearing-loss phenotypes are associated with NOG mutations than had previously been recognized, (2) patients with sporadic or familial nonsyndromic otosclerosis should be evaluated for mild features of this syndrome, and (3) NOG alterations should be considered in conductive hearing loss with subtle clinical and skeletal features, even in the absence of symphalangism.
Figures
References
Electronic-Database Information
-
- GenBank, http://www.ncbi.nlm.nih.gov/Genbank/ (for complete coding sequence [accession number U31202] and mRNA [accession number NM_005450])
-
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/ (for DFN3 [MIM 304400], NOG [MIM 602991], osteogenesis imperfecta type I [MIM 166200], otosclerosis [MIM 166800], stapes ankylosis with broad thumbs and toes [MIM 184460], SYM1 [MIM 185800], and SYNS1 [MIM 186500])
References
-
- Belecky-Adams T, Adler R (2001) Developmental expression patterns of bone morphogenetic proteins, receptors, and binding proteins in the chick retina. J Comp Neurol 430:562–572 - PubMed
-
- Brunet LJ, McMahon JA, McMahon AP, Harland RM (1998) Noggin, cartilage morphogenesis, and joint formation in the mammalian skeleton. Science 280:1455–1457 - PubMed
-
- Collins FS, Brooks LD, Chakravarti A (1998) A DNA polymorphism discovery resource for research on human genetic variation. Genome Res 8:1229–1231 - PubMed
-
- Gong Y, Krakow D, Marcelino J, Wilkin D, Chitayat D, Babul-Hirji R, Hudgins L, Cremers CW, Cremers FP, Brunner HG, Reinker K, Rimoin DL, Cohn DH, Goodman FR, Reardon W, Patton M, Francomano CA, Warman ML (1999) Heterozygous mutations in the gene encoding noggin affect human joint morphogenesis. Nat Genet 21:302–304 - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
