

Comparison of a new pmp22 transgenic mouse line with other mouse models and human patients with CMT1A
- PMID: 12090404
- PMCID: PMC1570695
- DOI: 10.1046/j.1469-7580.2002.00039.x
Comparison of a new pmp22 transgenic mouse line with other mouse models and human patients with CMT1A
Abstract
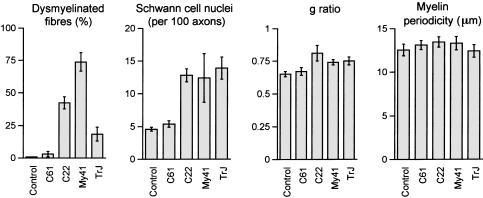
Charcot-Marie-Tooth disease type 1A is a dominantly inherited demyelinating disorder of the peripheral nervous system. It is most frequently caused by overexpression of peripheral myelin protein 22 (PMP22), but is also caused by point mutations in the PMP22 gene. We describe a new transgenic mouse model (My41) carrying the mouse, rather than the human, pmp22 gene. The My41 strain has a severe phenotype consisting of unstable gait and weakness of the hind limbs that becomes obvious during the first 3 weeks of life. My41 mice have a shortened life span and breed poorly. Pathologically, My41 mice have a demyelinating peripheral neuropathy in which 75% of axons do not have a measurable amount of myelin. We compare the peripheral nerve pathology seen in My41 mice, which carry the mouse pmp22 gene, with previously described transgenic mice over-expressing the human PMP22 protein and Trembler-J (TrJ) mice which have a P16L substitution. We also look at the differences between CMT1A duplication patients, patients with the P16L mutation and their appropriate mouse models.
Figures








References
-
- Ayers MM, Anderson RM. Onion bulb neuropathy in the Trembler mouse: a model of hypertrohic interstitial neuropathy (Dejerine-Sottas) in man. Acta. Neuropathol. 1973;25:54–70. - PubMed
-
- Baichwal RR, DeVries GH. A mitogen for Schwann cells derived from myelin basic protein. Biochem. Biophys. Res. Comm. 1989;164:883–888. - PubMed
-
- Berciano J, Garcia A, Calleja J, Combarros O. Clinico-electrophysiological correlation of extensor digitorum brevis muscle atrophy in children with Charcot-Marie-Tooth disease 1A duplication. Neuromusc. Disord. 2000;10:419–424. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
