

Structure of coronavirus main proteinase reveals combination of a chymotrypsin fold with an extra alpha-helical domain
- PMID: 12093723
- PMCID: PMC126080
- DOI: 10.1093/emboj/cdf327
Structure of coronavirus main proteinase reveals combination of a chymotrypsin fold with an extra alpha-helical domain
Abstract
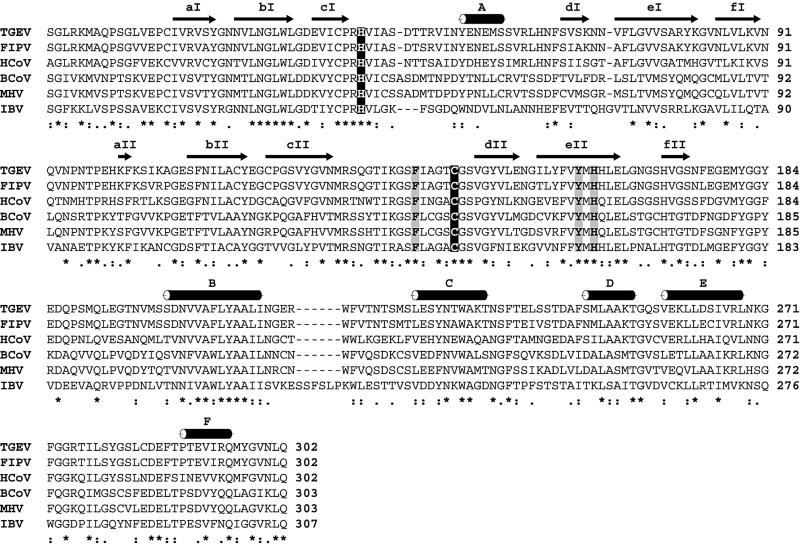
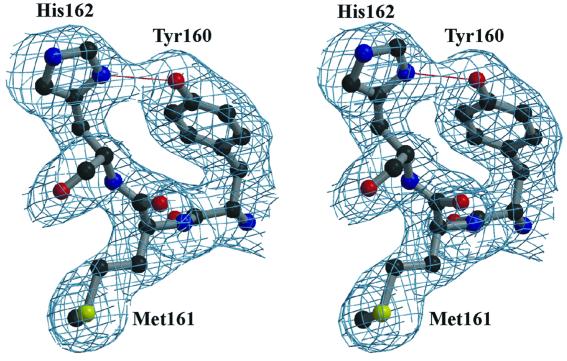
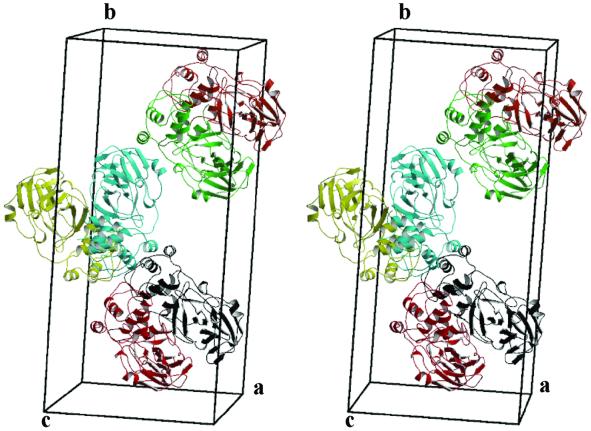
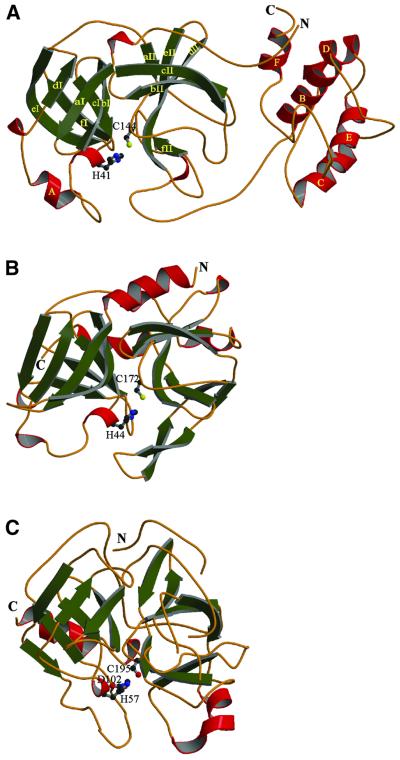
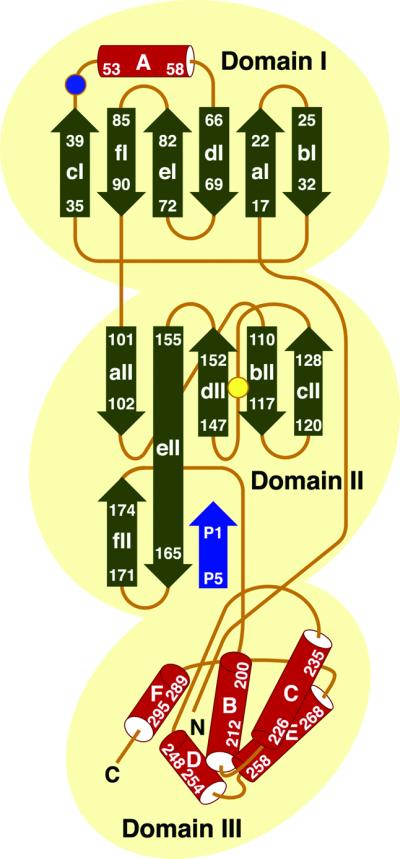
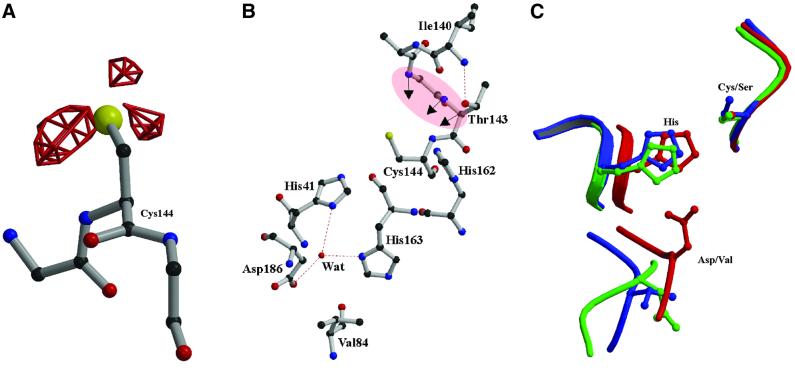
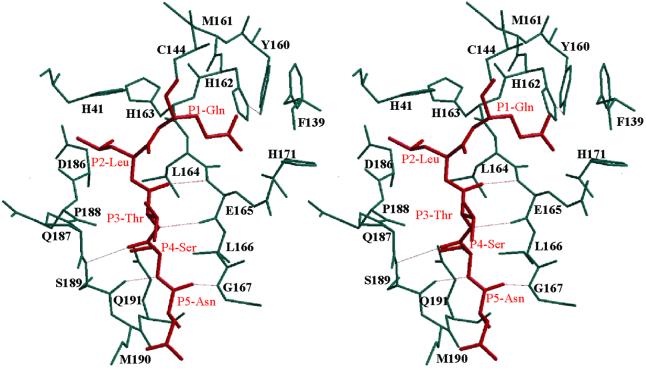
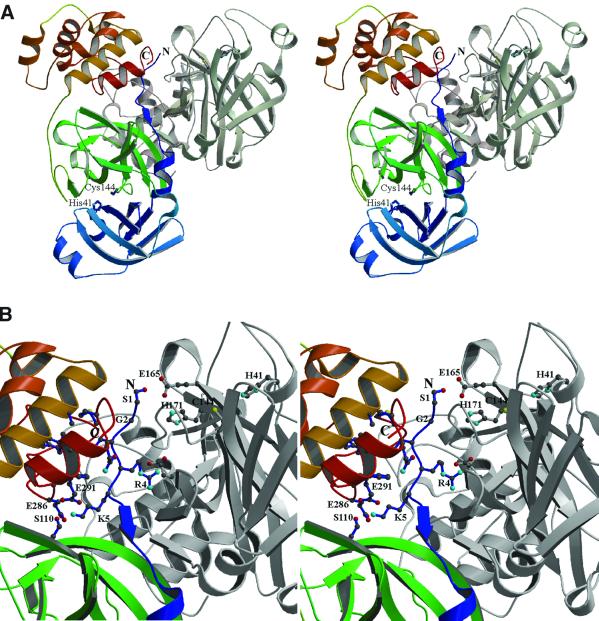
The key enzyme in coronavirus polyprotein processing is the viral main proteinase, M(pro), a protein with extremely low sequence similarity to other viral and cellular proteinases. Here, the crystal structure of the 33.1 kDa transmissible gastroenteritis (corona)virus M(pro) is reported. The structure was refined to 1.96 A resolution and revealed three dimers in the asymmetric unit. The mutual arrangement of the protomers in each of the dimers suggests that M(pro) self-processing occurs in trans. The active site, comprised of Cys144 and His41, is part of a chymotrypsin-like fold that is connected by a 16 residue loop to an extra domain featuring a novel alpha-helical fold. Molecular modelling and mutagenesis data implicate the loop in substrate binding and elucidate S1 and S2 subsites suitable to accommodate the side chains of the P1 glutamine and P2 leucine residues of M(pro) substrates. Interactions involving the N-terminus and the alpha-helical domain stabilize the loop in the orientation required for trans-cleavage activity. The study illustrates that RNA viruses have evolved unprecedented variations of the classical chymotrypsin fold.
Figures
References
-
- Allaire M., Chernaia,M.M., Malcolm,B.A. and James,M.N. (1994) Picornaviral 3C cysteine proteinases have a fold similar to chymotrypsin-like serine proteinases. Nature, 369, 72–76. - PubMed
-
- Bacon D.J. and Anderson,W.F. (1988) A fast algorithm for rendering space-filling molecule pictures. J. Mol. Graphics, 6, 219–220.
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
